From
PharmacotherapyTelaprevir: A Novel NS3/4 Protease Inhibitor for the Treatment of Hepatitis COlga M. Klibanov, Pharm.D.; Shannon H. Williams, Pharm.D.; Lisa S. Smith, Pharm.D.; Jacqueline L. Olin, M.S., Pharm.D.; Stephen B. Vickery
Posted: 10/26/2011; Pharmacotherapy. 2011;31(10):951-974. © 2011 Pharmacotherapy Publications
Abstract Hepatitis C virus (HCV) infection affects millions of people worldwide; however, standard therapy with peginterferon and ribavirin has resulted in suboptimal responses. Thus, new anti-HCV drugs with novel mechanisms of action are being studied. In particular, new drugs are being developed that target the NS3/4A protease complex. We evaluated the literature on telaprevir, a new, oral, covalent, reversible NS3/4A HCV protease inhibitor. A MEDLINE search (January 1996–July 2011) was performed to identify relevant clinical trials, and abstracts from hepatology and human immunodeficiency virus (HIV) conferences were reviewed. In large clinical trials, the addition of telaprevir to peginterferon and ribavirin resulted in high sustained virologic response rates in both treatment-naive and treatment-experienced patients infected with HCV genotype 1. Clinical data with telaprevir in the HIV-HCV coinfected population are emerging, as well as data on potential drug-drug interactions with this agent. Preliminary data describe the resistance profile of telaprevir; however, more information is needed in this evolving area. Telaprevir's most common adverse events included rash, pruritis, and anemia. Based on available data, this new anti-HCV drug will likely be widely used and may revolutionize the treatment of HCV-infected individuals.
Introduction
Hepatitis C virus (HCV) infection affects over 170 million people worldwide and is the most common blood-borne infection in the United States.[1, 2] Clinical trials with HCV therapy usually address four types of responses (Table 1), with sustained virologic response (SVR) being the most clinically important end point and goal of therapy for HCV-treated patients. At the time of this writing, the standard therapy for chronic HCV infection was the combination of peginterferon alfa and ribavirin.[3] This combination therapy results in approximately 55% of patients with HCV achieving SVR. The SVR rates are highest in patients with HCV genotypes 2 or 3 (70–80%), but low in patients with HCV genotype 1 (40–45%), patients coinfected with human immunodeficiency virus (HIV; 14–29%), African-Americans (19–23%), and patients with liver cirrhosis (~10%).[4–12] Because of the low response rates, the focus of clinical research for HCV infection has shifted toward direct-acting antivirals, including NS3/4 serine protease inhibitors and NS5B polymerase inhibitors (nucleoside and nonnucleoside).[13]
Final results of phase III clinical trials with the NS3/4 protease inhibitor telaprevir have been presented, and the manufacturer of telaprevir (Vertex Pharmaceuticals Inc., Cambridge, MA) submitted a new drug application to the United States Food and Drug Administration (FDA), seeking approval of telaprevir for the treatment of genotype 1 HCV, on November 23, 2010.[14] On May 23, 2011, the FDA approved telaprevir (Incivek) for the treatment of genotype 1 chronic HCV infection in adults with compensated liver disease, to be used in combination with peginterferon and ribavirin.[15]
In this review, we provide an overview of telaprevir, focusing on the drug's pharmacology, pharmacokinetics, pharmacodynamics, and the clinical data that led to its approval by the FDA. We performed a MEDLINE search (January 1996–July 2011) to identify relevant clinical trials and review articles. Abstracts and oral as well as poster presentations from meetings of the American Association for the Study of Liver Diseases, Conference on Retroviruses and Opportunistic Infections, and European Association for the Study of the Liver were also reviewed. Search terms were hepatitis C, directacting antivirals, NS3/4 serine protease inhibitors, telaprevir, and VX-950.
Pharmacology
Research on the life cycle of HCV has identified potential drug targets of antivirals that inhibit steps of HCV replication. Hepatitis C virus is a member of the Flaviviridae family of viruses and has a 9.6-kb positive-strand RNA genome that encodes a polyprotein precursor of approximately 3000 amino acids.[16–18] After attachment and fusion of the HCV to human hepatocytes, the viral nucleocapsid is released intracellularly (Figure 1). Uncoating of viral nucleocapsids in the cell cytoplasm then occurs, followed by synthesis of the HCV polyprotein. Hepatitis C virus genome translation at host ribosomes is controlled by the internal ribosome entry sites. The main role of these sites is to direct the virus to the endoplasmic reticulum. The polyprotein precursor encoded by HCV RNA is processed by both cellular and viral proteases to 10 individual proteins, including four structural proteins (C, E1, E2, and p7) and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B). Among the nonstructural proteins, NS5B RNA-dependent RNA polymerase and NS3, which consists of an N-terminal serine protease domain and a C-terminal helicase domain, are essential for translation and polyprotein processing; therefore, HCV replication can be blocked by NS3/4A protease inhibitors.
 | Figure 1. Replication of the hepatitis C virus (HCV) occurs in the following sequence: (a) receptor binding and cell entry of HCV into the hepatocyte; (b) uncoating and release of RNA; (c) the internal ribosome entry site directs the virus to the host endoplasmic reticulum; (d) translation and processing of polyprotein precursors requires viral NS4/NS4a protease; (e) transcription and development of progeny virions in capsomeres; and (f) newly formed virus released from hepatocyte cell membrane. (Adapted with permission from reference 13.) |
After posttranslational processing, HCV replication occurs. The exact mechanisms of HCV replication are not yet clearly defined but are thought to be dependent on the RNA-dependent RNA polymerase NS5B enzyme.[16–19] This enzyme has a catalytic site for nucleoside binding and at least four other sites for non-nucleoside binding, resulting in allosteric changes. The enzyme is a highly conserved structure across all HCV genotypes, which makes it ideal for drug targets. Several HCV polymerase inhibitors are under development. After replication, viral assembly occurs, which leads to transport and release into the extracellular space.[16–19]
Telaprevir is a small-molecule, peptidomimetic ketocarbonyl, covalent, reversible inhibitor of NS3/4A protease that was discovered by using a structure-based drug design approach.[20] The bond formation between telaprevir and NS3/4A protease has been described as a slow-on, slow-off relationship, with the enzyme-inhibitor complex half-life estimated to be 1 hour.[20] Studies evaluating telaprevir's antiretroviral antiviral activity were performed with HCV replicon cells and HCV-infected human fetal hepatocytes.[20] The steady-state inhibition constant was determined to be 7 nM. In these studies, the drug concentration required for 50% viral replication inhibition was 354 nM and 280 nM in the replicon cells and the fetal hepatocytes, respectively.
Pharmacokinetics and Pharmacodynamics
Preclinical studies with telaprevir investigated the drug's pharmacokinetic parameters in rats and dogs (Table 2).[20, 21] In one study, telaprevir had a moderate-to-high systemic clearance and a volume of distribution at steady state (Vdss) greater than total body water, suggesting good tissue distribution.[20] Its half-life in the animals ranged from 0.93–3.32 hours, suggesting that telaprevir should be dosed several times a day. Liver concentrations of telaprevir were also analyzed. A group of rats were given single doses of telaprevir 30 mg/kg in propylene glycol. The rats were sacrificed, and their livers were sliced into small pieces and homogenized with an equal volume of water. The drug concentrations were determined in liver and plasma by the achiral method. Telaprevir concentrations were higher in the liver relative to those in plasma at all time points tested (0.5, 1, 2, 4, and 8 hrs). The average liver concentration in the rats was 9.82 mg/g compared with the average plasma concentration of 0.28 mg/ml. Assuming the density of the liver was 1.0 g/ml, the liver:plasma concentration ratio in this study was 35:1, suggesting that telaprevir is a good candidate to treat HCV infection. A dose-dependent increase in liver and plasma drug concentrations was seen. At the lowest dose in animals (~10 mg/kg in dogs), the liver concentration of telaprevir was approximately 6-fold higher than the plasma concentration, supporting first-pass metabolism.
Human and rat liver microsomes have also been used to study telaprevir pharmacokinetics. After rats received a bolus of intravenous telaprevir 5 mg/kg, telaprevir had a half-life of 2.8 hours, volume of distribution (Vd) of 8.1 L/kg, and a Vdss of 2.4 L/kg (Table 2).[21] The discrepancy between Vd and Vdss may be explained by the binding of telaprevir to plasma proteins, blood cells, and other tissue components at steady state, resulting in a Vdss that is lower than the Vd. When telaprevir was given orally with ritonavir (a strong cytochrome P450 [CYP] 3A inhibitor), the maximum concentration (Cmax) increased from 0.43 μg/ml to 1.45 μg/ml, and the plasma concentration at 8 hours increased from 0.18 μg/ml to 0.97 μg/ml. Telaprevir's area under the concentration-time curve (AUC) increased from 1.05 μg•hr/ml to 8.86 μg•hr/ml, and its bioavailability was greater than 100%, suggesting that telaprevir is a substrate for CYP3A.
Pharmacokinetic parameters of telaprevir have also been studied in clinical trials (Table 3).[22, 23] In a phase Ib trial comparing three doses of telaprevir monotherapy versus placebo in 34 patients, the largest decline in HCV RNA corresponded with the highest mean trough level of telaprevir (1054 ng/ml) in the patients receiving telaprevir 750 mg every 8 hours (Table 3).[22] Patients receiving telaprevir 450 mg every 8 hours and those receiving 1250 mg every 12 hours had mean trough levels of 781 and 676 ng/ml, respectively. A correlation between trough concentrations and viral rebound was not found in these subjects. However, most patients receiving telaprevir 450 mg every 8 hours or 1250 mg every 12 hours had an HCV RNA rebound or plateau response, whereas patients receiving 750 mg every 8 hours had a continuous HCV RNA decline.
The two peginterferon products that are currently FDA approved for HCV therapy have different dosing recommendations and pharmacokinetic properties due to their difference in size, geometry, and attachment site in the polyethylene glycol moiety.[24] The recommended dosage for peginterferon alfa-2a is 180 μg/week, whereas the dosage of peginterferon alfa-2b is based on weight at 1.5 μg/kg/week.[3] Due to these differences, the pharmacokinetics of telaprevir were explored in a subset of 35 patients who were enrolled in a phase II trial with 161 total patients.[23] The study evaluated telaprevir 750 mg every 8 hours versus 1125 mg every 12 hours, and peginterferon alfa-2a versus peginterferon alfa-2b coadministration. At week 8, the mean Cmax of telaprevir, when given with peginterferon alfa-2a, was similar between the every-8-hour and every-12-hour dosing groups (4523 ng/ml and 4882 ng/ml, respectively; Table 3). In the every-12-hour group, the minimum concentration (Cmin) was 21% lower (least squares means [LSM] ratio 0.79, 90% confidence interval [CI] 0.63–0.99) and the AUC from 0–24 hours (AUC0–24) was 6% lower (LSM ratio 0.94, 90% CI 0.76–1.17) than those in the every-8-hour group. When coadministered with peginterferon alfa-2b, telaprevir's Cmax was 11% higher in the every-12-hour dosing group compared with the every-8-hour group (LSM ratio 1.11, 90% CI 0.97–1.27), and Cmin and AUC0–24 were comparable (90% CIs of LSM ratios were within 80–125%). The differences in the pharmacokinetic parameters in this trial did not translate into a difference in clinical outcome.
Clinical Trials of Efficacy and Toxicity
Telaprevir has been studied in several clinical trials for the treatment of HCV. A summary of those trials is presented in Table 4.
Phase I Trials
Telaprevir was initially studied in a phase Ib, randomized, dose-escalation, double-blind, placebo-controlled trial involving 34 patients with HCV genotype 1 infection.[22] The majority of patients (79%) had received previous treatment. Patients were administered 14 days of telaprevir 450 mg every 8 hours, 750 mg every 8 hours, or 1250 mg every 12 hours, or placebo. Although one patient discontinued the study drug due to an acute illness, and two withdrew their consent, data for all 34 patients were still reported. After 14 days, the placebo group experienced no change in their median HCV RNA level, whereas the three telaprevir dose groups (450 mg every 8 hours, 750 mg every 8 hours, and 1250 mg every 12 hours) experienced a median maximum decline of 3.46, 4.77, and 3.49 log10 IU/ml, respectively (statistical analysis not performed). All adverse events in the telaprevir-treated patients were of mild intensity, except for three events that were of moderate intensity. None of the moderate events were considered to be related to treatment with telaprevir. No clinically significant laboratory changes in the hematologic or metabolic panels were noted, as well as no clinically significant changes in electrocardiographic results.
After establishing the antiviral effects of telaprevir monotherapy, a phase I, randomized trial in 20 treatment-naive patients with HCV genotype 1 infection was conducted to establish the drug's antiviral activity when used in combination with peginterferon alfa-2a.[25] Patients were randomized into one of three 14-day treatment groups: placebo plus peginterferon alfa-2a, telaprevir monotherapy, or telaprevir plus peginterferon alfa-2a. Telaprevir was given as a 1250-mg loading dose followed by 750 mg every 8 hours, and peginterferon alfa-2a was dosed at 180 μg/week. All 20 patients completed the study. The median decrease in HCV RNA level from baseline to day 15 was 1.09 log10 IU/ml in the placebo plus peginterferon alfa-2a group, 3.99 log10 IU/ml in the telaprevir monotherapy group, and 5.49 log10 IU/ml in the telaprevir plus peginterferon alfa-2a group (statistical analysis not performed). Of the eight patients in the telaprevir monotherapy group, four were found to have viral variants conferring decreased sensitivity to telaprevir (low-level resistance: V36A/M and R155K/T; high-level resistance: A156V/T, V36A/M + R155K/T, or V36A/M + A156V/T). Of the eight patients who received telaprevir and peginterferon, two had viral variants resistant to telaprevir (one patient with A156T and one patient with V36A).
Adverse events were reported in all four patients (100%) in the placebo plus peginterferon alfa-2a group, six patients (75%) in the telaprevir monotherapy group, and all eight (100%) patients in the telaprevir plus peginterferon alfa-2a group. Adverse events that were moderate in severity and considered to be related to study drugs occurred in two patients in the telaprevir plus peginterferon alfa-2a group (headache, rash, insomnia) and in one patient in the telaprevir monotherapy group (pruritis). All other adverse events were of mild intensity. The most common events in the placebo plus peginterferon alfa-2a group, telaprevir monotherapy group, and telaprevir plus peginterferon alfa-2a group, respectively, were headache (50%, 25%, and 63%), myalgias (50%, 25%, and 63%), dry skin (25%, 25%, and 38%), nausea (25%, 13%, and 38%), and diarrhea (25%, 25%, and 25%).
Phase I trials showed that treatment with telaprevir, optimally dosed at 750 mg every 8 hours, resulted in significant decline in HCV RNA levels and led to early and more rapid HCV viral clearance compared with peginterferon alfa-2a therapy.
Phase II Trials
The first clinical trial to assess the antiviral activity and safety of telaprevir in combination with peginterferon alfa-2a and ribavirin was conducted in 12 treatment-naive adults with chronic HCV genotype 1 infection.[26] In this phase II, single-arm, open-label study, all patients received telaprevir as a 1250-mg loading dose followed by 750 mg every 8 hours, peginterferon alfa-2a 180 μg/week, and ribavirin 1000 mg/day (if weight < 75 kg) or 1200 mg/day (if weight ≥ 75 kg) for 28 days. After completing the 28-day study period, all patients were offered an additional 44 weeks of standard treatment (peginterferon alfa-2a + ribavirin). Due to the exploratory nature of the study, descriptive statistics were used, without any prospective calculation of statistical power. All 12 patients completed the trial. At the end of the study period (day 28), a decrease of at least 4 log10 IU/ml in HCV RNA levels was seen in all 12 patients, and 10 patients experienced a decrease of greater than 5 log10 IU/ml. Two patients experienced viral breakthrough during the peginterferon-ribavirin phase of treatment, and both were found to have telaprevir-resistant viral variant R155K.
All 12 patients reported at least one adverse event. Mild adverse events were noted in five patients (42%): five (42%) experienced influenza-like illness, four (33%) reported fatigue, and four (33%) complained of nausea. Anemia was noted in four patients (33%), which was also mild in severity. Adverse events of moderate severity were noted in six patients (50%): two (17%) had influenza-like illness, two (17%) reported fatigue, two (17%) developed a pruritic rash, and one (8%) had a headache. Only one patient (8%) reported a severe adverse event—headache.
Results from two large phase IIb trials with telaprevir in treatment-naive patients have been published.[27, 28] The Protease Inhibition for Viral Evaluation 1 (PROVE1) study was a randomized, double-blind, multicenter trial that enrolled 263 treatment-naive patients infected with HCV genotype 1 in the United States.[27] The study was designed to explore whether initiating therapy with telaprevir could shorten the duration of the current standard of care (48 wks). Study patients were randomized into one of four groups: the PR48 (control) group, in which 75 patients received placebo plus peginterferon alfa-2a plus ribavirin for 12 weeks, followed by peginterferon alfa-2a and ribavirin for 36 additional weeks; the T12PR24 group, in which 79 patients received telaprevir plus peginterferon alfa-2a and ribavirin for 12 weeks, followed by peginterferon alfa-2a and ribavirin for 12 more weeks; the T12PR48 group, in which 79 patients received telaprevir plus peginterferon alfa-2a plus ribavirin for 12 weeks, followed by peginterferon alfa-2a and ribavirin for 36 more weeks; and the T12PR12 group, in which 17 patients received telaprevir plus peginterferon alfa-2a plus ribavirin for 12 weeks. (This last group was an exploratory group with an intentionally smaller sample size.) Telaprevir was dosed as 1250 mg on day 1, followed by 750 mg every 8 hours; peginterferon alfa-2a was given as 180 μg/week; and ribavirin was given as 1000 mg/day (body weight < 75 kg) or 1200 mg/day (body weight ≥ 75 kg).
Several predefined stopping rules were implemented in this trial. In accordance with the standard of care,[3] therapy was discontinued in the PR48 group if HCV RNA levels did not decline by at least 2 log10 by week 12 or if the HCV RNA level was still detectable (> 10 IU/ml) at week 24. In the T12PR12 and T12PR24 groups, patients were required to have undetectable HCV RNA levels by week 4 (rapid virologic response [RVR]). During the treatment period, HCV RNA levels were measured on days 1 and 4, and at weeks 1, 2, 3, 4, 6, 8, 10, 12, 16, 20, 24, 28, 36, and 48. If the HCV RNA level was detectable at any time during weeks 4–20 (for the T12PR24 group) or weeks 4–10 (for the T12PR12 group), patients could not continue with their assigned duration of therapy (12 or 24 wks) and were switched to receive peginterferon and ribavirin for the standard total of 48 weeks. These patients were considered treatment failures in the analysis.
The primary end point of the PROVE1 study was the proportion of patients achieving SVR, based on an intent-to-treat analysis. The primary planned analysis was the comparison of the PR48 group with the T12PR24 group. Assuming a 50% SVR rate in the PR48 group and a 75% SVR rate in the T12PR24 group, 80 patients were needed in each group to provide 85% statistical power to show a statistical difference at a significance level of 5%, using a 2-sided t test.
Of the 263 randomized patients in the PROVE1 study, 250 received at least one dose of study drug. Of these patients, 26 (33%) of 79 in the T12PR24 group, 25 (32%) of 79 in the T12PR48 group, 4 (24%) of 17 in the T12PR12 group, and 17 (23%) of 75 in the PR48 group discontinued the study early. The study population was mostly Caucasian (77%) and male (63%), with a mean age of 48 years; 87% of the patients had a baseline HCV RNA level of greater than 800,000 IU/ml. The following SVR rates were seen in the study groups 24 weeks after the end of treatment: 41% in the PR48 group, 61% in the T12PR24 group (p=0.02 for the comparison with the PR48 group), 67% in the T12PR48 group (p=0.002 for the comparison with the PR48 group; p=0.51 for the comparison with the T12PR24 group), and 35% in the T12PR12 group (statistical analysis not performed for this group). In the small subgroup of African-American patients, only 11% (1 of 9) experienced SVR in the PR48 group compared with 44% (8 of 18) in the telaprevir groups. Of the 175 telaprevir-treated patients, 12 (7%) experienced viral breakthrough. Viral variants with telaprevir-associated mutations (V36M, R155K, A156T) were seen in 11 (92%) of the 12 patients (the remaining patient had wild-type virus due to several days of missed doses).
Most adverse events in the PROVE1 study were consistent with the usual interferon-induced systemic adverse effects. However, the following adverse events were reported more often in the telaprevir-treated patients than in the controls: rash (53–61% vs 41%), pruritis (24–48% vs 23%), nausea (48–65% vs 29%), and diarrhea (24–42% vs 28%). A greater proportion of patients discontinued treatment due to adverse events in the telaprevir groups compared with the control group (21% vs 11%). Serious adverse events occurred in 22 patients (4 in the control group and 18 in the telaprevir groups); 15 (68%) of the 22 events were considered to be related to study drug. Serious adverse events occurring in more than one patient occurred only in the telaprevir groups: three patients developed rash, three had anemia, three experienced ocular events (retinal detachment and scotoma), and two reported depression. The rashes were maculopapular and sometimes accompanied by pruritis. Laboratory changes were similar between groups, except for a decrease in hemoglobin level, which was more common in the telaprevir groups. Compared with the control group, the telaprevir groups experienced a 0.5–1-g/dl greater decline in hemoglobin level. Erythropoietin use was not permitted during the first 12 weeks of the study.
Unlike the PROVE1 trial, which recruited participants solely from the United States, the PROVE2 study was performed in Europe.[28] In this phase IIb, multicenter, randomized, partially double-blind, placebo-controlled trial, 334 patients infected with HCV genotype 1 were randomized to one of four treatment groups. The first three groups were identical to the PROVE1 trial: PR48 (control), T12PR24, and T12PR12. The fourth group, T12P12, received 12 weeks of only telaprevir and peginterferon alfa-2a, without ribavirin. Treatment of the PR48, T12PR12, and T12PR24 groups was double blinded through week 10. Treatment of the T12P12 group was not blinded. In addition to evaluating the efficacy of various durations of telaprevir-based therapies, this study also looked at the possibility of not including ribavirin in the combination therapy for HCV. Drug dosing was identical to the PROVE1 trial.
The predefined stopping rule for the PR48 group was identical to the rule in the PROVE1 trial. In the telaprevir groups, patients were required to have undetectable HCV RNA levels at the last study visit before the planned end of treatment (i.e., week 10 for the T12PR12 and T12P12 groups and week 20 for the T12PR24 group). If the HCV RNA level was detectable at the last study visit before the planned end of treatment, the patient was considered a treatment failure in the analysis and was switched to receive peginterferon and ribavirin for a total of 48 weeks.
The primary objective of the PROVE2 study was to compare SVR rates in the PR48 group with those in the T12PR12 and T12P12 groups combined. A secondary objective was to compare SVR rates in each of the telaprevir groups with the rate in the PR48 group. An intent-to-treat analysis was used. Assuming an SVR rate of 50% in the PR48 group and 70% in the combined T12PR12 and T12P12 groups, 80 patients were needed in the PR48 group and 160 patients were needed in the combined group to yield 80% statistical power.
Of the 334 randomized patients, 323 patients received at least one dose of study drug. However, 10 (12%) of 82 in the T12PR12 group, 20 (25%) of 81 in the T12PR24 group, 8 (10%) of 78 in the T12P12 group, and 32 (39%) of 82 in the PR48 group discontinued the study early. Similar to the PROVE1 study, the PROVE2 trial enrolled mostly Caucasian (91%), male (57%) patients, with a mean age of 45 years; 81% had a baseline HCV RNA level of 800,000 IU/ml or higher. Sustained virologic response was achieved in 46% of patients in the PR48 group compared with 48% of patients in the T12PR12 and T12P12 groups combined (p=0.89). However, when the data were analyzed based on the secondary objective, SVR rates were signifi-cantly higher in the T12PR24 group compared with the PR48 group (69% vs 46%, p=0.004), but not significantly higher in the T12PR12 group compared with the PR48 group (60% vs 46%, p=0.12). The T12P12 had a smaller proportion of patients achieving SVR compared with the PR48 group (36% vs 46%, p=0.20). A significantly higher proportion of telaprevir-treated patients achieved RVR and early virologic response compared with patients in the control group—two treatment end points that are often good predictors of SVR.
The necessity of including ribavirin with telaprevir and peginterferon was demonstrated in the PROVE2 trial. Not only did the SVR rates increase when ribavirin was included, but the rates of viral breakthrough and relapse were also significantly lower in the groups that received ribavirin. In the telaprevir-treated patients, viral breakthrough or relapse was seen in 41 (53%) of 78 patients in the T12P12 group, 12 (15%) of 81 patients in the T12PR24 group, and 20 (24%) of 82 patients in the T12PR12 group. Viral breakthrough and relapse were most likely due to emergence of telaprevir resistance, as a resistance analysis showed that 35 patients in the T12P12 group, 11 patients in the T12PR24 group, and 15 patients in the T12PR12 group had viral variants with resistance to telaprevir (mutations V36A/M, T54A, R155K/T, A156S).
Compared with the control group, telaprevir-treated patients were more likely to experience pruritis (51–63% vs 35%) and rash (44–49% vs 35%). The rash was maculopapular, and the median time to rash appearance was 9–12 days. Severe (grade 3) rash was seen in 13 (5%) of 241 patients treated with telaprevir but was not seen in any patient in the control group. Grade 4 rashes were not reported. Rash resulted in the discontinuation of treatment in 7% of telaprevir-treated patients. Anemia was also more common in the telaprevir groups (9–27% vs 17% in the control group). During the first 12 weeks of therapy, the median decline in hemoglobin level from baseline was 3.0 g/dl in the PR48 group, compared with 3.1 g/dl in the T12P12 group, 3.6 g/dl in the T12PR24 group, and 3.9 g/dl in the T12PR12 group. The use of erythropoietin was not allowed during the initial 12 weeks of the trial.
Although most of the available data with telaprevir is in the treatment-naive HCV-infected population, the drug has also been studied in treatment-experienced patients. The PROVE3 study was a phase IIb, randomized, stratified, partially placebo-controlled, partially double-blinded study that enrolled 465 patients in the United States, Canada, the Netherlands, and Germany with HCV genotype 1 infection who did not achieve a SVR after a full course of peginterferon and ribavirin combination therapy.[29] Previous nonresponse to standard therapy was defined as undetectable HCV RNA level never achieved during or at the end of treatment. Previous relapse was defined as undetectable HCV RNA level achieved during treatment for at least 42 weeks but detectable HCV RNA levels observed during the follow-up period and lack of achievement of SVR. Previous breakthrough was defined as undetectable HCV RNA level during the treatment period but detectable levels before the end of the treatment period.
Patients were randomized to one of four treatment groups: 115 patients received telaprevir plus peginterferon alfa-2a plus ribavirin for 12 weeks, followed by placebo plus peginterferon alfa-2a and ribavirin for the next 12 weeks (T12PR24); 113 patients received telaprevir plus peginterferon alfa-2a plus ribavirin for 24 weeks, followed by peginterferon alfa-2a and ribavirin for an additional 24 weeks (T24PR48); 111 patients received telaprevir plus peginterferon alfa-2a for 24 weeks (T24P24); and 114 patients received placebo plus peginterferon alfa-2a plus ribavirin for 24 weeks, followed by peginterferon alfa-2a and ribavirin for an additional 24 weeks (PR48 [control group]). Study drugs were dosed as follows: telaprevir 1125-mg loading dose, then 750 mg every 8 hours, peginterferon alfa-2a 180 μg/week, and ribavirin 1000 (weight < 75 kg) or 1200 mg/day (weight ≥ 75 kg).
There were four prespecified stopping rules in this trial. If a viral breakthrough—defined as an increase in the HCV RNA level of greater than 1 log10 or an HCV RNA level > 100 IU/ml from an undetectable level at a previous time point—was observed between weeks 4–24, all study drugs were discontinued. The second rule was for nonresponse at week 4, stating that by week 4 of treatment, patients in the PR48 group must have experienced at least a 1-log10 decline in HCV RNA level from baseline, and patients in the telaprevir groups must have had undetectable HCV RNA levels. The third stopping rule stated that if a nonresponse was observed at week 12 (defined as failure to achieve at least a 2-log10 decline in HCV RNA level), all study drugs were stopped. The fourth stopping rule was if patients in the T24PR48 and PR48 groups experienced a nonresponse at week 24 (defined as detectable HCV RNA level). If patients met any of the four stopping rules, they were considered nonresponders to treatment in the final analysis. Undetectable HCV RNA levels in this trial were defined as less than 10 IU/ml.
The primary end point was the proportion of patients achieving SVR, based on an intent-totreat analysis. Assuming a SVR rate of 20% in the control group and 45% in the telaprevir groups, 110 patients were needed in each group for the trial to have 90% statistical power to detect a significant difference between groups.
Of the 465 randomized patients, 453 received at least one dose of study drug. However, 29 (25%) of 116 patients in the T12PR24 group, 58 (50%) of 117 patients in the T24PR48 group, 53 (46%) of 115 patients in the T24P24 group, and 78 (67%) of 117 patients in the PR48 group discontinued the study prematurely. The majority of patients in this trial were men (68%), 89% were Caucasian, mean age was 51 years, 16% had cirrhosis, and 92% had HCV RNA levels of 800,000 IU/ml or higher at baseline. With regard to their response to previous peginterferon alfa and ribavirin therapy, 57% were nonresponders, 36% had a relapse, and 7% had a breakthrough. Sustained virologic response rates were higher in each of the telaprevir groups (51% in T12PR24, 53% in T24PR48, and 24% in the T24P24 group) compared with the PR48 group (14%; p<0.001, p<0.001, and p=0.02, respectively, for the T12PR24, T24PR48, and T24P24 groups). Patients who had a viral breakthrough or relapse with previous standard therapy had higher SVR rates in this trial compared with those who were nonresponders to previous treatment. Logistic regression analysis showed that SVR was significantly associated with randomization to the T12PR24 or T24PR48 group, an undetectable HCV RNA level during a previous period of standard treatment, and a low baseline HCV RNA level (< 800,000 IU/ml).
Similar to the PROVE2 study, the PROVE3 trial showed that ribavirin is essential in the combination therapy for HCV. In addition to achieving higher SVR rates, telaprevir-treated patients who also received ribavirin (the T12PR24 and T24PR48 groups) experienced lower relapse rates (30% and 13%, respectively) and lower viral breakthrough rates (13% and 12%, respectively) compared with the rates of 53% and 32%, respectively, in patients who did not receive ribavirin (the T24P24 group). Sequencing analysis in these patients revealed similar mutations at the NS3 amino acids that were seen in the PROVE1 and PROVE2 studies (positions 36, 54, 155, and 156), conferring resistance to telaprevir.
Most common adverse events were consistent with interferon-type symptoms, including influenza-like illness (25–32%) and fatigue (46–67%). Patients in the telaprevir groups were more likely than those in the PR48 group to experience a rash (41–60% vs 20%) and pruritis (34–44% vs 15%). Serious adverse events were more frequent in the telaprevir groups compared with the PR48 group (17–24% vs 11%). Anemia was also more common in the telaprevir groups (8–27% vs 8%) but resolved after drug discontinuation. Erythropoietin use was not permitted. Patients assigned to telaprevir were more likely to discontinue the study due to adverse events compared with patients assigned to the control group (50% vs 4%), with skin disorders being the most frequent cause of study discontinuation in the telaprevir groups.
All four phase II trials of telaprevir in patients infected with HCV genotype 1 used telaprevir dosed at 750 mg every 8 hours and combined telaprevir with peginterferon alfa-2a and ribavirin.[26–29] To investigate whether telaprevir can be administered every 12 hours and to explore its efficacy with peginterferon alfa-2b and ribavirin, an open-label, randomized, phase II trial was performed in 166 European treatment-naïve adults with HCV genotype 1 infection.[23] Patients were randomized to one of four treatment groups: group A received telaprevir 750 mg every 8 hours plus peginterferon alfa-2a plus ribavirin, group B received telaprevir 750 mg every 8 hours plus peginterferon alfa-2b plus ribavirin, group C received telaprevir 1125 mg every 12 hours plus peginterferon alfa-2a plus ribavirin, and group D received telaprevir 1125 mg every 12 hours plus peginterferon alfa-2b plus ribavirin. Standard subcutaneous doses of peginterferon alfa-2a (180 μg/wk) and peginterferon alfa-2b (1.5 μg/kg/wk) were used. The FDA-approved ribavirin dosing is weight based and differs depending on the peginterferon product used. This trial utilized these dosing guidelines, and depending on the patient's weight, ribavirin was given as 1000 or 1200 mg/day when administered with peginterferon alfa-2a and 800 or 1200 mg/day when administered with peginterferon alfa-2b. At randomization, patients were stratified based on their HCV subtype (1a or 1b) and baseline HCV RNA level (< 800,000 or ≥ 800,000 IU/ml).
All study patients received 12 weeks of their assigned telaprevir-based treatment, followed by peginterferon alfa and ribavirin alone for additional 12 or 36 weeks, based on their on-treatment virologic response. During the treatment period, HCV RNA levels were measured on days 1, 2, 3, 4, and 8, and at weeks 2, 3, 4, 6, 8, 10, 12, 14, 16, 20, 24, 36, and 48. If an undetectable HCV RNA level was achieved at weeks 4–20, a total of 24 weeks of therapy was given. If this criterion was not met, a total of 48 weeks of therapy was assigned. Additional treatment modifications were based on the following prespecified rules: if HCV RNA level was greater than 1000 IU/ml at weeks 4, 6, or 8, telaprevir was discontinued and peginterferon alfa and ribavirin were continued until week 48; if HCV RNA level did not decrease by more than 2 log10 from baseline by week 12 or if the HCV RNA level was detectable at week 24 or 36, then peginterferon alfa and ribavirin were also stopped.
The primary end point of the study was the proportion of patients achieving SVR. It was predetermined that if the interaction of telaprevir and peginterferon was not significant at the 10% level (i.e., the difference in SVR rates was less than 10%, irrespective of the peginterferon product used), the telaprevir and peginterferon groups would be pooled. To compare SVR rates, data were analyzed using a logistic regression model and adjusted for stratification factors. Due to the explorative nature of the trial, prospective power calculations were not performed, and statistical testing was performed at the 0.05 significance level without adjustment for multiple comparisons.
Of the 166 randomized patients, 161 received treatment and were included in the data analysis. Of these patients, 6 (15%) of 40 in group A, 8 (19%) of 42 in group B, 8 (20%) of 40 in group C, and 11 (28%) of 39 in group D discontinued the study prematurely. Fifty percent of the 161 patients were women, 90% were Caucasian, median age was 45 years, and 81% had a baseline HCV RNA level of 800,000 IU/ml or higher. Virologic response rates at week 4, week 12, and at the end of treatment were similar between all groups. The SVR rates were also similar in the four treatment groups: 85.0%, 81.0%, 82.5%, and 82.1% in groups A, B, C, and D, respectively (p>0.787). In the pooled telaprevir analysis, the SVR rates were 82.9% and 82.3% in the groups that received telaprevir every 8 hours and every 12 hours, respectively (p=0.997, 95% CI –11.4–11.4%). In the pooled peginterferon analysis, the SVR rates were 83.8% and 81.5% in the patients receiving alfa-2a and alfa-2b, respectively (p=0.906, 95% CI –10.8–12.1%). In the 161 patients who received treatment, viral relapse and breakthrough were observed in 9 (6%) and 14 (9%) patients, respectively. Viral sequence analyses showed viral mutations consistent with previously reported data,[20, 23, 24] at NS3 positions 36, 54, 155, and 156.
The adverse events were similar between the four treatment groups. Serious adverse events were noted in 20 (12.4%) of the 161 patients, most frequently anemia in five patients (3.1%) and rash in four patients (2.5%). Severe (grade 3) rash was seen in six patients (3.7%). Study drugs were discontinued in 13 patients (8.1%) due to adverse events; seven patients (4.3%) discontinued mainly due to rash and four patients (2.5%) discontinued due to anemia. Grade 3 anemia—defined as a hemoglobin level less than 8.0 g/dl in females or less than 8.5 g/dl in males—was seen in 19 patients (11.8%). This study allowed erythropoietin use, and it was prescribed in 25% of patients. This was the first clinical trial that compared both of the licensed peginterferon alfa plus ribavirin treatments in combination with the same NS3/4A protease inhibitor, showing that SVR rates were similar, irrespective of the peginterferon product used. This was also the first clinical trial to show that SVR rates did not differ between the every-8-hour and the every-12-hour telaprevir dosing regimen. Although these results are promising, they should be interpreted with caution due to the exploratory nature of the study, the relatively small sample size, and the lack of a control group. The study was also open label, not statistically powered, and enrolled only a European population without any cirrhosis at baseline. The applicability of these results to other patient populations is therefore unknown, and the effectiveness of the treatment strategies used in this study should be confirmed with further clinical data.
Most of the phase II data with telaprevir in the treatment-naive population have been obtained in patients infected with HCV genotype 1. Some initial data with genotypes 2, 3, and 4 have recently become available.[30, 31] A phase IIa, partially blinded, randomized study (study C209) of 49 treatment-naive patients infected with HCV genotypes 2 and 3 was conducted.[30] Patients were randomized to one of three groups: 10 patients with genotype 2 and eight with genotype 3 received telaprevir alone (group A), five patients with genotype 2 and nine with genotype 3 received telaprevir plus peginterferon alfa-2a and ribavirin (group B), and eight patients with genotype 2 and nine with genotype 3 received peginterferon alfa-2a and ribavirin plus placebo (group C). All patients received their specified treatment for 14 days, followed by peginterferon and ribavirin through week 24, which is the standard duration of treatment for patients with genotypes 2 and 3. Dosages were as follows: telaprevir 750 mg every 8 hours, peginterferon alfa-2a 180 μg/week, and ribavirin 800 mg/day. The primary end point was HCV RNA level decline at day 15. An intent-to-treat analysis was utilized when all treated patients completed 15 days of dosing or discontinued study drugs earlier than day 15.
In patients infected with HCV genotype 2, the mean log10 decrease in HCV RNA level at day 15 was 3.66, 5.51, and 4.83 for groups A, B, and C, respectively. The SVR was achieved in 56%, 100%, and 89% of patients, respectively. Given the quick development of resistance to NS3/4A inhibitors, it is surprising that the SVR rate in group A is as high as 56%. The short 14-day duration of telaprevir treatment may not have had a significant impact on the efficacy of the remaining 22 weeks of peginterferon plus ribavirin therapy. These results should be interpreted with caution due to the very small sample size in each study group.
In patients infected with HCV genotype 3, the mean log10 decrease in HCV RNA level at day 15 was 0.54, 4.85, and 4.72 for groups A, B, and C, respectively. The SVR was achieved in 50%, 67%, and 44% of patients, respectively. These efficacy results were also surprising, given that previous studies have shown SVR rates as high as 80% with standard peginterferon and ribavirin therapy in patients infected with HCV genotype 3.[4, 5] Only 26 patients infected with HCV genotype 3 were studied in this trial; thus, the results need to be validated in larger clinical trials.
The overall frequency of adverse events was similar in the groups. The most common adverse events in the telaprevir-treated patients were rash, nausea, and flu-like symptoms. One patient in group B discontinued the study due to rash. No serious adverse events were reported.
Telaprevir has also been studied in 24 treatment-naïve patients with HCV genotype 4, in a phase IIa, randomized, partially blinded trial (study C210).[31] Patients were randomized to receive 2 weeks of either telaprevir alone (eight patients), telaprevir plus peginterferon alfa-2a and ribavirin (eight patients), or placebo plus peginterferon alfa-2a and ribavirin (eight patients). Telaprevir was dosed as 750 mg every 8 hours, peginterferon alfa-2a as 180 μg/week, and ribavirin as 1000 or 1200 mg/day. After the initial 14 days of the study, all patients received standard of care (peginterferon plus ribavirin) for 46 additional weeks. Because this was the first study to evaluate telaprevir in patients with HCV genotype 4 infection, the investigators aimed to quantify the antiviral activity of the drug, and the primary end point was the HCV RNA level decrease at day 15. The more clinically important end point, SVR rates, was a secondary end point. An intent-to-treat analysis was used when all treated patients completed 15 days of study or discontinued the study earlier than day 15.
The greatest HCV RNA level decrease at day 15 was seen in the telaprevir plus peginterferon plus ribavirin group (–4.32 log10), compared with the telaprevir alone group (–0.77 log10) and the placebo plus peginterferon and ribavirin group (–1.58 log10). This early decline in the HCV RNA level in the telaprevir plus peginterferon plus ribavirin group did not translate to improved SVR rates following a subsequent 46 weeks of standard therapy; SVR rates were 63%, 50%, and 63% of patients in the telaprevir alone, telaprevir plus peginterferon plus ribavirin, and placebo plus peginterferon plus ribavirin groups, respectively. The overall frequency of adverse events was similar across study groups. One patient experienced a serious adverse event that was considered to be unrelated to telaprevir but led to treatment discontinuation.
Data from phase II studies of telaprevir suggest that this drug, when combined with peginterferon and ribavirin, improves SVR rates in patients infected with HCV genotype 1. The data from the larger phase II trials (PROVE1 and PROVE2) also suggest that the duration of therapy can possibly be reduced from 48 weeks to 24 weeks for many treatment-naive patients when telaprevir is combined with standard therapy. The PROVE3 study showed that when patients who previously did not achieve a SVR with standard peginterferon and ribavirin therapy are retreated with telaprevir plus peginterferon plus ribavirin, high SVR rates are achieved in this difficult-to-treat population. The PROVE3 study also compared 12 versus 24 weeks of telaprevirbased initial therapy, showing that SVR rates were similar between these two groups, but premature study discontinuations and rates of adverse events were lower in the group that received only 12 initial weeks of telaprevir. These efficacy and safety data suggest that using telaprevir for 12 weeks results in a better risk-benefit profile than using telaprevir for 24 weeks.
The inclusion of ribavirin in the combination therapy for HCV is important, as its inclusion increased SVR rates, prevented relapse, and prevented the emergence of telaprevir resistance. Results from smaller phase II trials also suggest that although telaprevir may have significant antiviral activity against HCV genotype 2,[30] its activity against HCV genotypes 3 and 4 may be limited,[30, 31] supporting the need for additional studies with this agent in patients infected with HCV that is not genotype 1.
One of the main limitations to the phase II trials is the underrepresentation of difficult-totreat patient populations (African-Americans, patients with cirrhosis, those coinfected with HIV), so the applicability of results to these patient populations is unknown. In addition, although the every-12-hour dosing strategy in one study showed similar SVR rates with an every-8-hour dosing strategy,[23] this should be investigated further, since any dosing techniques to improve adherence will likely play a major role in the treatment of HCV. Several of the phase II trials[26, 30, 31] enrolled a very small number of study patients and were, therefore, not statistically powered, which makes it difficult to interpret their results. Nevertheless, data from phase II clinical trials of telaprevir were promising.
Phase III Trials
At the time of this writing, final results of two phase III trials of telaprevir in treatment-naive patients infected with HCV genotype 1 were available (Table 4).[33, 34] The ILLUMINATE trial was a phase III, randomized, open-label study that enrolled 540 treatment-naive patients with genotype 1 HCV infection, with or without cirrhosis, at 74 sites in the United States.[33] The main objectives of this trial were to evaluate the safety and efficacy of two treatment durations of telaprevir with peginterferon and ribavirin.
During the first 12 weeks all study patients received telaprevir 750 mg every 8 hours plus peginterferon alfa-2a 180 μg/week and ribavirin 1000 or 1200 mg/day. Peginterferon alfa-2a with ribavirin alone were given during weeks 12–20. If patients discontinued treatment for any reason before week 20, they were included in the "early discontinuation" group. Rapid virologic response was defined as an undetectable HCV RNA level (< 25 IU/ml) at week 4. Extended rapid virologic response (eRVR) was defined as an undetectable HCV RNA level (< 25 IU/ml) at weeks 4 and 20. At week 20, those patients who had an eRVR were randomized (in a 1:1 ratio) to continue peginterferon and ribavirin until week 24 (T12PR24 group) or until week 48 (T12PR48 group). All patients without eRVR continued peginterferon and ribavirin until week 48. The SVR rates were measured 24 weeks after completion of therapy in all groups.
Several prespecified stopping rules were in place. Patients discontinued telaprevir and continued peginterferon and ribavirin if their HCV RNA level was greater than 1000 IU/ml at week 4. All study drugs were stopped if the HCV RNA level did not decrease by at least 2 log10 from baseline to week 12 and if the HCV RNA level was still detectable between weeks 24 and 36. If patients experienced anemia, their ribavirin dosage was reduced, but erythropoietin use was not permitted. The primary end point was the proportion of patients achieving SVR; the secondary end point was safety. This trial had a noninferiority design to evaluate a 24-week versus 48-week duration of treatment in patients who achieved eRVR, with a predefined noninferiority margin of –10.5%. An intent-to-treat analysis was used and included all randomized patients.
One hundred patients discontinued treatment before week 20 and thus were included in the early discontinuation group. The rates of premature study discontinuation in patients with eRVR who were randomized were as follows: 1 (0.6%) of 162 patients in the T12PR24 group and 26 (16%) of 160 patients in the T12PR48 group. Of the patients without eRVR at week 20, 33 (28%) of 118 patients discontinued the study prematurely. The ILLUMINATE trial enrolled predominantly male (60.2%), Caucasian (79.1%) patients, whose median age was 51 years and median baseline HCV RNA level was 6.5 log10 IU/ml; 11.3% of patients had bridging fibrosis or compensated cirrhosis at study entry.
Of the 540 patients in the intent-to-treat population, 389 (72%) achieved RVR and 352 (65.2%) achieved eRVR. Treatment duration of 24 weeks was noninferior to 48 weeks in patients who achieved eRVR (SVR rates: 92% vs 87.5%, difference of 4.5%, 95% CI –2.1–11.1% [2-sided t test]). The SVR rate was 64% in the group of patients without eRVR and 23% in the early discontinuation group. The overall SVR in the intent-to-treat analysis was 71.9%. High SVR rates were also seen in difficult-to-treat populations. Of the 149 patients with liver fibrosis at baseline, 94 (63%) achieved SVR. A high SVR rate of 60% (44 patients) was also noted in the 73 African-American patients.
The most common adverse events, occurring in at least 25% of patients, included fatigue, pruritis, nausea, anemia, headache, rash, insomnia, diarrhea, and flu-like symptoms. Of the 540 patients, 94 (17.4%) discontinued all study drugs permanently due to adverse events. During the first 12 weeks of telaprevir, 3 (0.6%) and 6 (1.1%) patients, respectively, discontinued treatment due to anemia and rash.
Final results of the ADVANCE trial have been published.[34] This phase III, randomized, double-blind, multicenter, placebo-controlled trial conducted in the United States and Europe enrolled 1095 treatment-naive patients with HCV genotype 1 infection. The objective of this trial was to evaluate a response-guided approach to determine the duration of HCV treatment. Patients were randomized to one of three treatment groups (in a 1:1:1 ratio): telaprevir plus peginterferon alfa-2a plus ribavirin administered for 8 weeks, followed by additional weeks of peginterferon alfa-2a and ribavirin; telaprevir plus peginterferon alfa-2a plus ribavirin administered for 12 weeks, followed by additional weeks of peginterferon alfa-2a and ribavirin; and placebo plus peginterferon alfa-2a plus ribavirin administered for 12 weeks, followed by 36 weeks of peginterferon alfa-2a and ribavirin (control group). Telaprevir was dosed as 750 mg every 8 hours, peginterferon alfa-2a as 180 μg/week, and ribavirin as 1000 or 1200 mg/day. Definitions of RVR and eRVR were identical to the ILLUMINATE trial.[33] Patients in the telaprevir groups achieving eRVR received a total of 24 weeks of therapy whereas those who did not achieve eRVR received a total of 48 weeks of treatment.
Prespecified stopping rules were similar to the ILLUMINATE trial.[33] Telaprevir was stopped and peginterferon and ribavirin continued if a patient's HCV RNA level was greater than 1000 IU/ml at week 4. All study drugs were discontinued if the HCV RNA level did not decrease by at least 2 log10 from baseline to week 12 and if the HCV RNA level was still detectable between weeks 24–40. If patients experienced anemia, their ribavirin dosage was decreased, but erythropoietin use was not allowed. The primary end point, based on an intent-to-treat analysis, was SVR rates.
In the intent-to-treat population (1088 patients), the rates of premature study discontinuation were as follows: 95 (26%) of 363 patients in the 12-week telaprevir group, 104 (29%) of 364 patients in the 8-week telaprevir group, and 159 (44%) of 361 patients in the control group. The study population of 1088 patients was 58% men and 88% Caucasian. The median age was 49 years, 77% of patients had an HCV RNA level of 800,000 IU/ml or higher at baseline, and 21% had bridging fibrosis or compensated cirrhosis. Significantly higher RVR rates were seen in the telaprevir-treated groups compared with the control group (66% in the 8-wk telaprevir group vs 68% in the 12-wk telaprevir group vs 9% in the control group). Nearly 60% of telaprevir-treated patients achieved an eRVR and were assigned to the short duration of therapy (24 wks). The SVR rates were 69% in the 8-week telaprevir group, 75% in the 12-week telaprevir group, and 44% in the control group (p<0.0001 for each telaprevir group vs the control group). The overall rate of virologic failure was lower with 12 weeks of telaprevir (8%) compared with 8 weeks (13%). Higher SVR rates were seen with telaprevir in patients with no or minimal or portal fibrosis (73% in the 8-wk telaprevir group, 78% in the 12-wk telaprevir group, and 47% in the control group) as well as patients with bridging fibrosis or cirrhosis (53% in the 8-wk telaprevir group, 62% in the 12-wk telaprevir group, and 33% in the control group). African-American patients also experienced higher SVR rates if they received telaprevir (58% in the 8-wk telaprevir group, 62% in the 12-wk telaprevir group, and 25% in the control group).
Similar to the ILLUMINATE trial,[33] the most common (> 25% of patients) adverse events in all groups were fatigue, pruritis, nausea, headache, anemia, rash, influenza-like illness, insomnia, pyrexia, and diarrhea. Pruritis, rash, nausea, anemia, and diarrhea occurred more frequently in the telaprevir groups than in the control group.
Whereas the ILLUMINATE[33] and ADVANCE[34] trials were performed in treatment-naive HCV-infected patients, the REALIZE study was a phase III, randomized, international, double-blind, placebo-controlled study that enrolled 663 patients infected with HCV genotype 1 who were treatment experienced.[35] Patients were eligible to enroll in this trial if they had a nonresponse (i.e., null or partial response) or a relapse with previous peginterferon and ribavirin therapy. Patients were randomized in a 2:2:1 ratio to receive telaprevir plus peginterferon alfa-2a and ribavirin for 12 weeks, followed by peginterferon alfa-2a and ribavirin for 36 weeks (T12PR48); peginterferon alfa-2a and ribavirin for 4 weeks, followed by telaprevir plus peginterferon alfa-2a and ribavirin for 12 weeks, then peginterferon alfa-2a and ribavirin for 32 weeks (lead-in T12PR48); or placebo plus peginterferon alfa-2a and ribavirin for 48 weeks (PR48 [control]). Study dosages were telaprevir 750 mg every 8 hours, peginterferon alfa-2a 180 μg/week, and ribavirin 1000 or 1200 mg/day.
The primary objective of this study was to evaluate SVR rates in the telaprevir groups for patients with a history of nonresponse or relapse to previous therapy. Secondary objectives included the evaluation of a lead-in phase, and SVR rates with telaprevir in previous null and partial responders. The SVR was defined as an undetectable (< 25 IU/ml) plasma HCV RNA level 24 weeks after the last dose of study drug.
In the intent-to-treat population (662 patients), the rates of premature study discontinuation were as follows: 100 (38%) of 266 patents in the T12PR48 group, 79 (30%) of 264 patients in the lead-in T12PR48 group, and 82 (62%) of 132 patients in the PR48 group. At baseline, 70% of patients were male, 93% were Caucasian, 26% had cirrhosis, and 89% had a baseline HCV RNA level of 80,000 IU/ml or higher. Overall, 53% of the patients had a previous relapse, 19% had a partial response, and 28% had no response. In previous relapsers, the SVR rate in the control group (PR48) was 24%, compared with 83% in the T12PR48 group (p<0.0001 vs PR48), and 88% in the lead-in T12PR48 group (p<0.001 vs PR48). In previous nonresponders, the SVR rate in the control group (PR48) was 9%, compared with 41% in the T12PR48 group (p<0.0001 vs PR48), and 41% in the lead-in T12PR48 group (p<0.001 vs PR48). In prior partial responders, the SVR rate in the control group (PR48) was 15%, compared with 59% in the T12PR48 group (p<0.0001 vs PR48), and 54% in the lead-in T12PR48 group (p<0.001 vs PR48). In previous null responders, the SVR rate in the control group (PR48) was 5%, compared with 29% in the T12PR48 group (p<0.0001 vs PR48), and 33% in the lead-in T12PR48 group (p<0.001 vs PR48). A lead-in phase did not have a significant impact on SVR rates in this trial.
The adverse events in the REALIZE trial were similar to those reported in the phase III ADVANCE[34] and ILLUMINATE[33] trials. Adverse events occurring in more than 25% of patients included fatigue, pruritis, headache, rash, nausea, flu-like symptoms, anorectal symptoms, insomnia, diarrhea, pyrexia, cough, and asthenia. Adverse events that were more commonly seen in telaprevirtreated patients compared with controls included fatigue, pruritis, rash, nausea, anemia, anorectal symptoms, and diarrhea. The most common reasons for study discontinuation secondary to adverse effects were rash (4%) and anemia (3%). Erythropoietin use was not permitted.
Several subanalyses from the REALIZE study have been reported.[36, 37] One subanalysis evaluated patients' responses at week 4 of treatment and their association with SVR, focusing on the telaprevir delayed start group (lead-in T12PR48).[36] It was found that although there were similar percentages of patients at week 4 with a less than 1-log10 decline in both the lead-in T12PR48 group and the PR48 group, the percentage of SVR was higher in the lead-in T12PR48 group (62% in relapsers, 56% in partial responders, and 15% in null responders) compared with the PR48 group (0%).
Due to previous studies suggesting that a polymorphism near the IL28B gene is associated with increased SVR rates in patients treated with peginterferon and ribavirin,[38, 39] a retrospective analysis of the REALIZE study was performed to evaluate the SVR rates in patients with the IL28B genotype.[37] Eighty percent of patients consented to genetic testing, and 18% of patients were IL28B C/C, 61% C/T, and 21% T/T. In this analysis, IL28B genotype did not help predict outcomes in previous treatment-experienced patients, so the utility of this testing remains unknown in this setting.
Phase III clinical trials of telaprevir in treatment-naïve and treatment-experienced patients infected with HCV genotype 1 confirmed the clinical benefit of telaprevir that had been demonstrated in phase II studies. In treatment-naïve patients, the data suggest that after the initial 12 weeks of treatment with telaprevir, peginterferon and ribavirin, 12 additional weeks of peginterferon and ribavirin (total of 24 wks of therapy) result in higher SVR rates than the current standard of therapy. The data from the ADVANCE trial[34] also suggest that a longer initial duration of 12 weeks with telaprevir plus peginterferon plus ribavirin results in lower virology failure rates compared with 8 weeks.
Unlike clinical trials with another NS3/4A protease inhibitor, boceprevir, which suggest that an initial lead-in phase with peginterferon and ribavirin plays an important role in decreasing rates of virologic breakthroughs and drug resistance,[40–42] the ADVANCE trial did not show a significant benefit of the lead-in phase before telaprevir initiation.[34] Considering that the ADVANCE trial did not enroll any treatment-naïve patients, and that this was the only study with telaprevir, to our knowledge, that evaluated the lead-in phase, additional data are needed to discern whether or not this treatment strategy is beneficial with telaprevir.
Phase III trials of telaprevir also addressed difficult-to-treat HCV-infected populations. Data from the REALIZE study[35] showed that telaprevir has significant efficacy in these patients, especially those who experienced a partial response or a relapse to their prior regimen of peginterferon and ribavirin. Unlike phase II trials of telaprevir, the phase III studies allowed enrollment of patients with some level of liver cirrhosis. Previous trials with peginterferon and ribavirin reported extremely poor SVR rates of approximately 10% in this patient population;[12] however, patients with liver cirrhosis who received telaprevir as part of their HCV therapy achieved surprisingly high rates of SVR of up to 73% in the ILLUMINATE and ADVANCE trials.[33, 34] In addition, SVR rates up to 62% were seen in treatment-naïve African-American patients randomized to telaprevir[33, 34]—results that are extremely promising, considering that historically, SVR rates in African-American patients have been only 19–23% with the standard peginterferon and ribavirin treatment.[10, 11]
Although the phase III studies of telaprevir 750 mg every 8 hours showed excellent SVR rates, it would be of great interest to see phase III study results of treatment with telaprevir 1250 mg every 12 hours. At the time of this writing, none of the phase III studies evaluated this dosage, possibly because only the 750 mg every 8 hours dosage was studied in the larger phase II studies (PROVE 1[27] and PROVE2[28]). Decreasing the dosing frequency of telaprevir would likely play an important role in improving patient adherence as well as acceptability of the drug by patients. Other limitations to the phase III trials of telaprevir include underrepresentation of certain populations. Although SVR rates seemed very promising in African-Americans and in patients with cirrhosis, these patients represented a very small proportion of the study cohorts, and these results, therefore, should be interpreted with caution. In addition, the majority of the study patients were men, so the data with favorable SVR rates may not be applicable to female patients. All of the phase III trials excluded patients coinfected with HIV and HCV, and the high burden of HCV disease in the HIV-infected population makes this area of research extremely interesting to clinicians who treat these patients.
Human Immunodeficiency Virus–Hepatitis C Virus Coinfected Patients
Approximately 25–30% of HIV-infected persons in the United States are also infected with HCV.[43, 44] In this population, SVR rates are reported to be only 14–29% with standard therapy, which are significantly lower compared with the SVR rates of 40–80% in HIV-negative persons.[4–9] Due to the burden of HCV disease in the HIV-infected population and the historically low response rates in this patient population, information on new treatment for patients coinfected with HIV and HCV has been highly anticipated. At the time of this writing, only interim results from one clinical trial with HIV-HCV coinfected patients were available. A phase IIa, randomized, placebo-controlled, multicenter, two-part clinical trial (study 110) evaluated the clinical efficacy and safety of telaprevir plus peginterferon alfa-2a and ribavirin in patients coinfected with HIV and HCV genotype 1; patients could take efavirenz-based or boosted atazanavir-based antiretroviral therapy (Table 4).[32] In this two-part study, 60 patients took peginterferon alfa-2a and ribavirin for 48 weeks (control group) or telaprevir plus peginterferon alfa-2a plus ribavirin for 12 weeks, followed by peginterferon alfa-2a and ribavirin for an additional 36 weeks. Part A of the study included patients who were not receiving antiretroviral therapy. These patients were required to have a CD4+ cell count of 500 cells/mm3 or greater and an HIV-1 RNA level of 100,000 copies/ml or lower. Part B included patients who were receiving stable antiretroviral therapy (tenofoviremtricitabine-efavirenz or atazanavir-ritonavir plus tenofovir plus emtricitabine [or lamivudine]). Patients in part B were required to have a CD4+ cell count of 300 cells/mm3 or greater and an HIV-1 RNA level of 50 copies/ml or lower. Telaprevir was dosed as 750 mg every 8 hours; the dose was increased to 1125 mg every 8 hours in patients who were also receiving efavirenz. Peginterferon alfa-2a was given as 180 μg/week. The ribavirin dose differed depending on the location of the study center: it was weight based in France and Germany (1000 mg/day if weight < 75 kg; 1200 mg/day if weight ≥ 75 kg) and 800 mg/day at all other institutions.
Predefined stopping rules included discontinuation of telaprevir and continuation of peginterferon and ribavirin if a patient's HCV RNA level was 1000 IU/ml or higher at weeks 4 or 8. All study drugs for HCV were discontinued in all other viral breakthrough scenarios. The primary end points, based on an intent-to-treat analysis, were the proportion of patients with undetectable (< 25 IU/ml) HCV RNA levels at week 12 and safety. The secondary end points included the proportion of patients with an undetectable HCV RNA level at week 4, SVR at week 24 after the final telaprevir dose, HCV resistance, pharmacokinetics of telaprevir, and pharmacokinetics of antiretroviral agents in patients enrolled in part B of the study.
Interim results for 59 of the 60 patients (13 in part A and 46 in part B) have been presented. Of the 59 patients, most were male (86%) and Caucasian (68%); approximately 8% of study patients had bridging fibrosis at baseline. At week 12, the combined results (all patients receiving telaprevir) showed that 68% of patients in the telaprevir plus peginterferon alfa-2a and ribavirin groups achieved an undetectable HCV RNA level, compared with 14% of patients in the peginterferon alfa-2a and ribavirin groups (statistical analysis not reported). At week 4, an undetectable HCV RNA level was achieved in 70% of patients in the telaprevir plus peginterferon alfa-2a and ribavirin groups, compared with 5% of patients in the peginterferon alfa-2a and ribavirin groups. Results at weeks 4 and 12 were similar whether or not patients were on concurrent antiretroviral therapy. A decline in the HIV-1 RNA level of approximately 1 log10 copies/ml was seen by week 12 of HCV therapy in patients who were not receiving antiretroviral therapy (part B). Due to the preliminary nature of these data, the clinical significance of this decline in the HIV RNA level is not known. The data regarding an association between HCV infection and HIV disease progression are conflicting. Many studies have shown a direct negative impact of HCV infection on progression of HIV disease, whereas other trials do not support this relationship.[45–49] Longer follow-up in coinfected patients is needed to more clearly discern the clinical significance of lowering HIV RNA levels in HIV-coinfected patients who are not receiving antiretroviral therapy.
Adverse events that were more commonly seen in the telaprevir-treated patients compared with controls included nausea (35% vs 14%), pruritis (35% vs 5%), dizziness (22% vs 5%), and fever (22% vs 9%). Moderate-severity rashes were observed in 11% of patients receiving telaprevir compared with less than 1% of patients receiving peginterferon alfa-2a and ribavirin alone. Serious adverse events were reported in one patient receiving telaprevir but no antiretroviral therapy and in two patients receiving telaprevir and atazanavir-based antiretroviral therapy.
Plasma telaprevir trough concentrations were similar in the groups with and without antiretroviral therapy. Median concentrations of efavirenz, atazanavir, and tenofovir decreased by approximately 20% from baseline after initiation of telaprevir with peginterferon alfa-2a and ribavirin; the clinical significance of this is unknown. This is the first clinical trial that is evaluating HCV NS3/4 serine protease inhibitors in HIV-HCV coinfected patients. Compared with historical data in this patient population, the interim results from this trial are encouraging and should be validated with longer follow-up and additional studies.
Resistance
Hepatitis C virus has a very high viral replication rate coupled with an error-prone RNA-dependent RNA polymerase. For this reason, HCV exists in humans as wild-type and quasispecies virus (mutated virus). Quasispecies are rapidly selected with HCV protease inhibitor monotherapy, and a small percentage of viral variants are resistant to these drugs before any drug exposure. A study of 507 treatment-naive patients infected with HCV genotype 1 reported the frequency of patients with at least one dominant direct-acting antiviral resistance mutation was 8.6% for genotype 1a and 1.4% for genotype 1b.[50]
Viral resistance produced by NS3/4 protease inhibitor monotherapy is rapid. In a 14-day, phase Ib, randomized, double-blind, placebo-controlled trial of 34 patients with chronic HCV genotype 1, telaprevir monotherapy resulted in a 2–3-log10 decrease in HCV viral load within 2 days, but resistance began to develop thereafter.[22] Only 25% of patients had continued viral decline. Several single or double mutations were observed (V36A/M, T54A, R155K/T, and A156V/T/S) within 7–10 days after telaprevir administration and even 3–7 months afterward.
Low-level resistant variants are slower to be replaced by wild-type virus. A subanalysis of patients not achieving SVR in the ADVANCE, ILLUMINATE, and REALIZE studies followed patients for a median of 11 months after treatment failure to assess how long viral variants persisted after treatment failure with telaprevir.[51] The median time to loss of detectable T54A/S and A156S/T/V viral variants was 4 months. The V36A/M, R155I/K/M/T, and V36M + R155K variants were the slowest to be replaced by wildtype virus, with the median time of 10, 11, and 14 months, respectively. After 11 months, 60% of the patients had no detectable telaprevir-resistant variants, suggesting that viral resistance may resolve over time for many patients and they may be able to use other direct-acting antivirals in the future. These results should be interpreted with caution, however, given that in the field of HIV, all viral variants are archived by the virus.[52] Although the drug mutations may not be detectable under lack of drug pressure, the variants reemerge once the drug is restarted.
Research in the field of HIV has shown that certain mutations in the protease gene are selected for only by certain HIV protease inhibitors. However, considerable overlap does exist between the combinations of mutations in HIV strains that develop resistance to HIV protease inhibitors, which results in the wide cross-resistance that is typically observed within this class of antiretrovirals.[52] Unlike in the field of HIV, studies investigating cross-resistance between HCV NS3/4 protease inhibitors are sparse. In the in vitro studies of telaprevir and another NS3/4 protease inhibitor, boceprevir, there was cross-resistance in the A156T/V variant, but viral replication was still reduced. Both antiviral agents remained sensitive to peginterferon alfa-2a and ribavirin.[53–55]
Although our knowledge of resistance to the direct-acting antivirals is continuously evolving, initial data with telaprevir and other NS3/4 serine protease inhibitors suggest that resistance to these agents may occur pretreatment and that monotherapy with these drugs leads to rapid emergence of viral mutations. Further data on cross-resistance between these agents are needed to elucidate whether patients can take one of the drugs if they are resistant to the others. Of importance, the telaprevir-resistant virus that rapidly develops during monotherapy appears to remain sensitive to peginterferon and ribavirin.[53–58]
Drug-drug Interactions
A phase I, randomized trial in 20 patients administered telaprevir 1250 mg on day 1 followed by 750 mg every 8 hours examined the drug's effect on HCV RNA level changes when given with peginterferon alfa-2 180 μg/week and also evaluated the pharmacokinetics of both drugs.[25] On day 1, the Cmax of telaprevir was 3695 ng/ml when given alone and 3392 ng/ml when administered with peginterferon alfa-2a (p=0.81). A trend toward a higher telaprevir Cmax at steady state was seen when given with peginterferon alfa-2a, but the difference was not statistically significant.
In rat and human liver microsomes, the metabolism of telaprevir was strongly inhibited by the presence of ritonavir (a potent CYP3A inhibitor), suggesting that telaprevir is a substrate of CYP3A.[21] This route of metabolism would likely lead to potential drug-drug interactions between telaprevir and many antiretroviral agents that are also metabolized by CYP3A. Telaprevir's manufacturer has undertaken an extensive drug-drug interaction program with telaprevir, and although at the time of this writing, most of the data have only been described in telaprevir's package insert,[59] several drug-drug interaction studies have been presented at scientific conferences.[60–63]
An open-label, multiple-dose, crossover study in 24 healthy adults receiving telaprevir 750 mg every 8 hours and esomeprazole 40 mg once/day showed that the coadministration of the proton pump inhibitor did not have an effect on the pharmacokinetics of telaprevir.[60] These data suggest that the absorption of telaprevir is unaffected by changes in gastric pH and the drug can be safely administered with gastric acid suppressants such as antacids, histamine2-receptor blockers, and proton pump inhibitors.
Depression is a common adverse event in patients receiving peginterferon and ribavirin, and antidepressants are often used in patients receiving HCV therapy.[3] An open-label, randomized, multiple-dose, crossover pharmacokinetic study in 16 healthy adults examined the pharmacokinetics of escitalopram 10 mg/day and telaprevir 750 mg every 8 hours administered together. The study found no significant changes in the pharmacokinetic parameters of telaprevir.[61] Concentrations of escitalopram, however, decreased in the presence of telaprevir. The LSM ratios for the escitalopram Cmin, Cmax, and AUC0–24 were 0.58 (90% CI 0.52–0.64), 0.70 (90% CI 0.65–0.76), and 0.65 (90% CI 0.60–0.70), respectively. The mechanism of this interaction is not well understood but may be explained by telaprevir's induction of CYP2C19 or CYP3A, which are the major pathways of metabolism for escitalopram. The decrease of 35% of escitalopram exposure suggests that more aggressive dosing of select serotonin reuptake inhibitors may be needed in patients who are also receiving telaprevir.
Due to the high burden of HCV disease in HIVinfected patients, drug-drug interactions between telaprevir and antiretrovirals must be identified. A pharmacokinetic study of 18 healthy adult volunteers evaluated telaprevir pharmacokinetics when given with ritonavir (Table 5).[62] Participants were randomized to receive telaprevir 750 mg every 8 hours alone, telaprevir 750 mg every 12 hours plus ritonavir 100 mg every 12 hours, or telaprevir 250 mg every 12 hours plus ritonavir 100 mg every 12 hours. The mean Cmax and Cmin of telaprevir, when given alone as 750 mg every 8 hours, were 2808.17 ng/ml and 1386.17 ng/ml, respectively. When telaprevir was administered as 250 mg every 12 hours together with ritonavir 100 mg every 12 hours, the mean Cmax and Cmin of telaprevir were 1202.83 ng/ml and 352.33 ng/ml, representing a 59% decline in the Cmax and a 75% decline in the Cmin concentrations. Coadministration of telaprevir 750 mg every 12 hours with ritonavir 100 mg every 12 hours also resulted in lower concentrations of telaprevir, although not as pronounced as with the lower telaprevir dose. This combination resulted in the mean telaprevir Cmax of 2368 ng/ml and Cmin of 1386 ng/ml, representing a 15% decrease in the Cmax and a 32% decrease in the Cmin concentrations.
These results were surprising, as ritonavir is typically a strong inhibitor of CYP3A. The investigators hypothesized that self-inhibition of CYP3A-based metabolism by telaprevir may explain the lack of significant "boosting" by ritonavir. Other explanations are the induction of CYP3A and/or P-glycoprotein by ritonavir, or displacement of protein binding of telaprevir by ritonavir. Ritonavir exposure was higher when coadministered with telaprevir, suggesting some dose-dependent CYP3A inhibition by telaprevir. Most HIV regimens that include an HIV protease inhibitor use ritonavir, taking advantage of its CYP inhibitory properties and thereby decreasing the amount of the protease inhibitor needed to suppress HIV replication and decreasing the dosing frequency. However, the pharmacokinetic study with telaprevir and ritonavir coadministration[62] proved that this concept of boosting does not apply to this drug combination and that this strategy should not be employed to decrease the dosing frequency of telaprevir.
Three separate open-label, randomized, crossover studies were conducted in healthy volunteers to investigate the pharmacokinetics of telaprevir when administered with various antiretroviral agents (Table 5).[63] Coadministration of telaprevir 750 mg every 8 hours with lopinavir 400 mg–ritonavir 100 mg every 12 hours in 21 healthy volunteers resulted in a telaprevir AUC over the dosing interval (AUC0–τ) LSM ratio of 0.46 (90% CI 0.41–0.56) and telaprevir Cmin LSM ratio of 0.48 (90% CI 0.40–0.56), indicating a significant decrease in telaprevir exposure. Lopinavir concentrations did not change significantly, indicated by an AUC0–τ LSM ratio of 1.06 (90% CI 0.96–1.17) and Cmin LSM ratio of 1.14 (90% CI 0.96–1.36).
The same cohort of 21 healthy subjects received telaprevir 750 mg every 8 hours with atazanavir 300 mg–ritonavir 100 mg once/day.[63] Coadministration of these agents resulted in a telaprevir AUC0–τ LSM ratio of 0.80 (90% CI 0.76–0.85) and telaprevir Cmin LSM ratio of 0.85 (90% CI 0.75–0.98). In the presence of telaprevir, atazanavir AUC0–τ LSM ratio was 1.17 (90% CI 0.97–1.43) and atazanavir Cmin LSM ratio was 1.85 (90% CI 1.40–2.44).
Telaprevir 750 mg every 8 hours was also studied with darunavir 600 mg–ritonavir 100 mg every 12 hours in a pharmacokinetic study in 20 healthy volunteers.[63] Similar to the trial with lopinavir-ritonavir and atazanavir-ritonavir, telaprevir exposure also decreased in this trial. Coadministration of darunavir-ritonavir resulted in telaprevir AUC0–τ LSM ratio of 0.65 (90% CI 0.61–0.69) and Cmin LSM ratio of 0.68 (90% CI 0.63–0.74). Darunavir concentrations also declined, with the resulting AUC0–τ LSM ratio of 0.60 (90% CI 0.57–0.63) and Cmin LSM ratio of 0.58 (90% CI 0.52–0.63).
The combination of fosamprenavir 700 mg–ritonavir 100 mg every 12 hours with telaprevir 750 mg every 8 hours was also studied in the same cohort of 20 healthy volunteers.[63] A decrease in telaprevir exposure was noted, with a telaprevir AUC0–τ LSM ratio of 0.68 (90% CI 0.63–0.72) and a Cmin LSM ratio of 0.70 (90% CI 0.64–0.77). Fosamprenavir concentrations also declined, with a resulting AUC0–τ LSM ratio of 0.53 (90% CI 0.49–0.58) and a Cmin LSM ratio of 0.44 (90% CI 0.40–0.50).
These initial pharmacokinetic data suggest that telaprevir exposure is decreased in the presence of all ritonavir-boosted HIV protease inhibitors, with atazanavir-ritonavir having the lowest impact on telaprevir exposure. Telaprevir also has variable effects on the exposures of antiretrovirals, increasing the AUC of atazanavir and lopinavir, and decreasing the AUC of darunavir and fosamprenavir. The clinical consequences of these interactions are not known; however, based on these data, the United States product labeling for telaprevir recommends that telaprevir should not be given with the following HIV protease inhibitors: darunavir-ritonavir, fosamprenavirritonavir, or lopinavir-ritonavir.[59]
Efavirenz is a commonly prescribed nonnucleoside reverse transcriptase inhibitor that is a substrate as well as an inducer of CYP.[64] Thus, a drug-drug interaction between telaprevir and efavirenz is possible, necessitating higher doses of telaprevir when used with efavirenz. One study showed a decrease in telaprevir AUC and Cmin when telaprevir was taken with efavirenz (Table 5).[63] Coadministration of telaprevir 1125 mg every 8 hours with efavirenz 600 mg/day and tenofovir 300 mg/day resulted in the telaprevir AUC0–τ LSM ratio of 0.82 (90% CI 0.73–0.92) and a Cmin LSM ratio of 0.75 (90% CI 0.66–0.86). Changing the telaprevir dosage to 1500 mg every 12 hours did not improve telaprevir concentrations; the resulting telaprevir AUC0–_ LSM ratio was 0.80 (90% CI 0.73–0.88) and the Cmin LSM ratio was 0.52 (90% CI 0.42–0.64). Efavirenz and tenofovir concentrations remained relatively unchanged. Although the clinical implications of this interaction are not known, a higher dose of telaprevir, 1125 mg every 8 hours, is being studied in patients coinfected with HIV and HCV who are also receiving efavirenz-based antiretroviral therapy.[32]
Due to potential or documented significant drug-drug interactions, coadministration of the following drugs with telaprevir is contraindicated: α1-adrenergic antagonists, rifampin, ergot derivatives, cisapride, St. John's wort, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (atorvastatin, lovastatin, and simvastatin), pimozide, phosphodiesterase type 5 inhibitors (sildenafil or tadalafil when used for portal hypertension), midazolam, and triazolam.[59] Clinicians should be aware that many other drugs also have the potential to interact with telaprevir, especially if they are metabolized by the CYP or p-glycoprotein systems. The product labeling for telaprevir provides additional information on these drug-drug interaction and guidelines on how to manage them.
Dosing and Administration
Telaprevir is available as a 375-mg tablet. The recommended dose is 750 mg 3 times/day (doses taken 7–9 hrs apart) with food.[59] Telaprevir should always be administered with both peginterferon alfa and ribavirin for the first 12 weeks of HCV therapy. After the first 12 weeks, response-guided therapy should be implemented. Treatment-naive patients infected with HCV who achieved an undetectable HCV RNA level at weeks 4 and 12 of treatment should receive an additional 12 weeks of peginterferon alfa and ribavirin, and those who did not achieve an undetectable HCV RNA level at weeks 4 and/or 12 should receive peginterferon alfa and ribavirin for an additional 36 weeks.[59] Treatment-experienced patients should receive triple therapy with telaprevir, peginterferon alfa, and ribavirin for the first 12 weeks, followed by an additional 36 weeks of peginterferon alfa and ribavirin.[59]
It is important to note that the dosing strategy with telaprevir differs from the dosing strategy of another NS3/4A protease inhibitor recently approved by the FDA, boceprevir, due to the different dosing strategies employed with the two drugs in clinical trials. Unlike telaprevir, boceprevir should be started only after 4 weeks of peginterferon and ribavirin therapy, and the duration of boceprevir follows a different response-guided schedule.[65]
Telaprevir has been studied in several specific patient populations.[59] The steady-state exposure of telaprevir was 46% lower in HCV-negative patients with moderate hepatic impairment (Child-Pugh class B) compared with healthy volunteers; therefore, telaprevir should not be used in this patient population. Sex, race-ethnicity, and age (> 65 yrs old) did not affect the pharmacokinetics of telaprevir. The drug has not been studied in pediatric patients
Conclusion
The worldwide burden of HCV infection is significant. Until now, treatment with peginterferon and ribavirin for HCV has resulted in suboptimal responses. Data from landmark clinical trials with telaprevir have led to its FDA approval. Compared with standard 48-week peginterferon and ribavirin therapy, significantly higher SVR rates were achieved in patients infected with HCV genotype 1 when telaprevir was part of their treatment. Not only were SVR rates significantly improved, but the duration of therapy was also decreased to 24 weeks in treatment-naïve patients who achieved undetectable HCV RNA levels at weeks 4 and 12. A total of 48 weeks of treatment should be given to treatment-naïve patients if they do not experience an undetectable HCV RNA level at weeks 4 and 12.
Even treatment-experienced patients with HCV genotype 1, who typically have limited HCV retreatment options, benefited from triple therapy with telaprevir, peginterferon, and ribavirin, with up to 88% of patients achieving SVR in the phase III REALIZE trial. Unlike the treatment-naive population, however, treatment-experienced patients should receive a total of 48 weeks of treatment.
Limitations of adding telaprevir to standard treatment (peginterferon and ribavirin) include the increased risk of adverse effects, specifically rash, pruritis, and anemia. In addition, the added pill burden and dosing schedule may impact patients' adherence levels, leading to treatment failure. Limiting the duration of initial therapy with telaprevir, peginterferon, and ribavirin to only 12 weeks (and then continuing with peginterferon and ribavirin dual therapy) will be important due to the high risk of adverse events and cumbersome dosing schedule for telaprevir (every 8 hrs).
In this new era of direct-acting antivirals, clinicians will have many opportunities to be actively involved in managing HCV-infected individuals due to the numerous drug-drug interactions, complicated dosing schedules, and the high frequency of adverse drug reactions. Intensive adherence counseling will play a major role in the treatment of HCV, and treatment protocols will need to be constructed to manage adverse events. Despite these issues that may complicate treatment, telaprevir is one of the first direct-acting antivirals to treat HCV and will likely play an important role in revolutionizing HCV infection therapy.


 -defensin (HBD)-2 plays a role in host defense against the cold virus, too.
-defensin (HBD)-2 plays a role in host defense against the cold virus, too.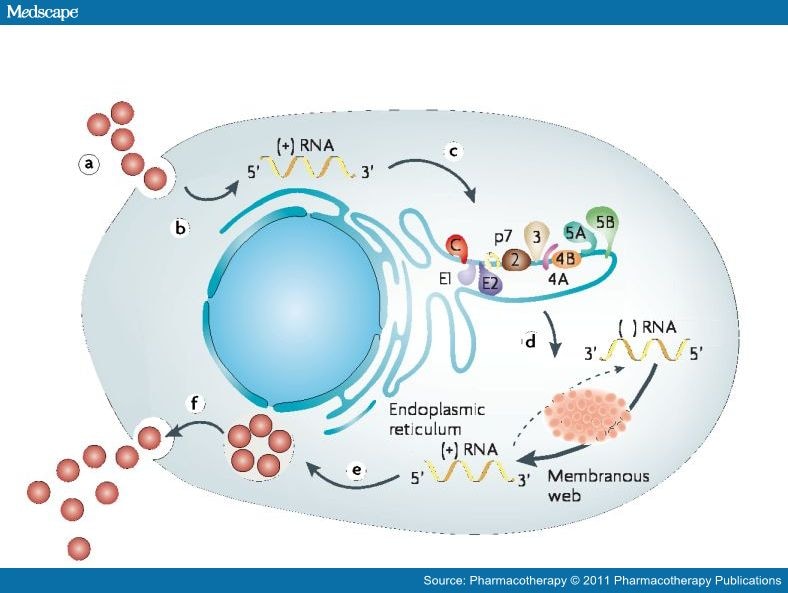{kind=link}