Recent advances in basic science
Interferon lambdas: the next cytokine storm
OPEN ACCESS
Christabel Kelly,
Paul Klenerman,
Eleanor Barnes
+ Author Affiliations
Abstract Full text PDF
Peter Medawar Building for Pathogen Research and Oxford NIHR Biomedical Research Centre, Nuffield Department of Medicine, University of Oxford, Oxford, UK
Correspondence to Dr Eleanor Barnes, Peter Medawar Building for Pathogen Research, Oxford NIHR Biomedical Research Centre, Nuffield Department of Medicine, University of Oxford, South Parks Road, Oxford OX1 3SY, UK; ellie.barnes@ndm.ox.ac.uk
Abstract
For two decades the scientific community has sought to understand why some people clear hepatitis C virus (HCV) and others do not. Recently, several large genome-wide association studies have identified single nucleotide polymorphisms (SNPs) linked to interferon lambda 3 (IFNλ3) that are associated with the spontaneous resolution and successful treatment of HCV infection. These observations are generating intense research activity; the hope is that IFNλ3 genetic variants may serve as important predictive biomarkers of treatment outcome and offer new insights into the biological pathways involved in viral control.
A pharmacogenomic treatment approach for HCV can now be envisaged, with the incorporation of host genetic variants into a predictive treatment algorithm with other factors. The SNPs associated with the clinical outcome of HCV infection are located some distance from the IFNλ3 gene itself, and causal genetic variants have yet to be clearly defined. Locating these causal variants, mapping in detail the IFNλ3 signalling pathways and determining the downstream genetic signature so induced will clarify the role of IFNλ3 in the pathogenesis of HCV. Clinical studies assessing safety and efficacy in the treatment of HCV with exogenous IFNλ3 are currently underway. Early results suggest that IFNλ3 treatment inhibits HCV replication and is associated with a limited side effect profile. However, hepatotoxicity in both healthy volunteers and HCV-infected patients has been described.
This review discusses the genetic studies that link IFNλ3 to both the spontaneous resolution and treatment-induced clearance of HCV and the potential impact of this in clinical practice, the biology of IFNλ3 as currently understood and how this may impact on HCV infection, and describes the early studies that assess the role of this cytokine in the treatment of patients with HCV.
Introduction
For more than two decades, interferon-α (IFNα) treatment has formed the cornerstone of therapy for hepatitis C virus (HCV) infection. Despite the identification of some viral and host factors associated with viral clearance, response to therapy remains highly unpredictable. Since treatment is prolonged, expensive and fraught with side effects, intense efforts have been made to identify biomarkers predictive of a successful treatment outcome. In 2009 there was a major breakthrough in this endeavour with the discovery that single nucleotide polymorphisms (SNPs) linked to the cytokine IFNλ3 (also known as IL28B) are a dominant host factor at a population level in determining treatment outcome. Furthermore, the same SNPs are associated with the clinical outcome of primary infection—an intense area of research in the field of HCV over recent years. It is therefore not surprising that IFNλ is creating a storm in the HCV arena.
This review describes the discovery of IFNλ3-linked SNPs that are associated with HCV control, summarises the biology and function of IFNλ3 as currently understood and describes the early studies that assess the role of this cytokine in the treatment of patients.
Identification of IFNλ3 as a key cytokine in HCV infection using genetic technology
Over the last decade a number of host factors have been shown to play a role in the clinical outcome of HCV using either a candidate gene approach or through the assessment of biological pathways or demographic features that have an expected role in infection control. For example, in acute infection, particular human leucocyte antigens (both HLA classes I and II) and robust T cell immunity have been associated with viral clearance.1–3
Single-source HCV outbreak studies most clearly demonstrate the importance of host factors in response to HCV infection.4 In these studies, young women were infected with the same inoculum of HCV from contaminated anti-D immunoglobulin. Approximately half of the women developed persistent infection while the remainder cleared the virus spontaneously, showing that host factors are important in determining clinical outcome. Similarly, in the treatment of persistent HCV infection, some host factors such as male gender and African ethnicity are associated with a poor treatment outcome.5–7 However, the actual predictive value of these factors is low, suggesting that other factors, hitherto unidentified, must exist. These observations provided the rationale for fully assessing the contribution of the host genome to the outcome of HCV infection.
The development of the human HapMap project in 2002 (http://hapmap.ncbi.nlm.nih.gov/) and advances in genetic techniques that facilitate the analysis of large datasets has led to an exponential growth in genome-wide association studies (GWAS). These studies associate common SNPs throughout the host genome with a disease cohort, and so identify important biological pathways in disease without prior knowledge or prejudice as to their relevance. This technique contrasts with candidate gene studies that take a pre-identified relevant gene and specifically assess its prevalence in the target population. The polarised clinical outcome of HCV infection (that of viral clearance versus viral persistence) makes HCV infection an ideal candidate for a GWAS approach.
The first GWAS in HCV infection was published by Ge and colleagues in September 2009.8 This study assessed the treatment outcome in a group of 1671 patients of mixed ethnicity (American European, Hispanic and African) receiving pegylated-IFNα (PEG- IFNα) and ribavirin. A striking association was discovered between sustained viral response (SVR) with treatment and a cluster of seven SNPs linked to the IL28B gene, with the most significant SNP (rs12979860) demonstrating extremely high statistical significance (p=1.06×10-25). No SNPs linked to other genes were associated with treatment outcome at genome-wide significance. Patients homozygous for the protective allele (C/C) had an SVR rate that was approximately three times higher than homozygotes for the risk allele (T/T) (78% vs 28%), and simply being a heterozygote carrier (T/C) for the risk allele also demonstrated approximately a twofold decrease in SVR (38%) compared with those homozygous for the protective allele, suggesting a dominant role for the risk SNP. In assessing other factors linked to viral clearance, host IFNλ3 genotype was more important than baseline viral load, the degree of liver fibrosis or ethnicity (figure 1).
{kind=link}
Odds ratio (OR) of treatment failure in hepatitis C virus (HCV) infection. OR of risk factors associated with treatment failure including the interferon λ3 (IFNλ3)-linked single nucleotide polymorphisms (rs8099917, rs12979860, rs12980275) in published genome-wide association studies.8–,11 *Derived from Rauch et al.9
Many other studies have since replicated these findings, demonstrating the robust association between IFNλ3 and treatment outcome in other populations including Japanese, Australian, European, African American and Hispanic using either a GWAS approach8–11 or a candidate gene approach (table 1, figures 2 and 3).12–18
{kind=link}
{kind=link}
Comparison of genome-wide association studies (GWAS) identifying single nucleotide polymorphisms (SNPs) linked to interferon λ3 (IFNλ3) associated with treatment response or spontaneous clearance
Interferonλ3 (IFNλ3) risk allele frequency in specific populations infected with hepatitis C virus (HCV). Risk allele frequency in those who clear acute infection spontaneously and in those who have a successful treatment response (SVR) compared with individuals who develop persistent infection or who fail treatment (non-SVR).8–11 18

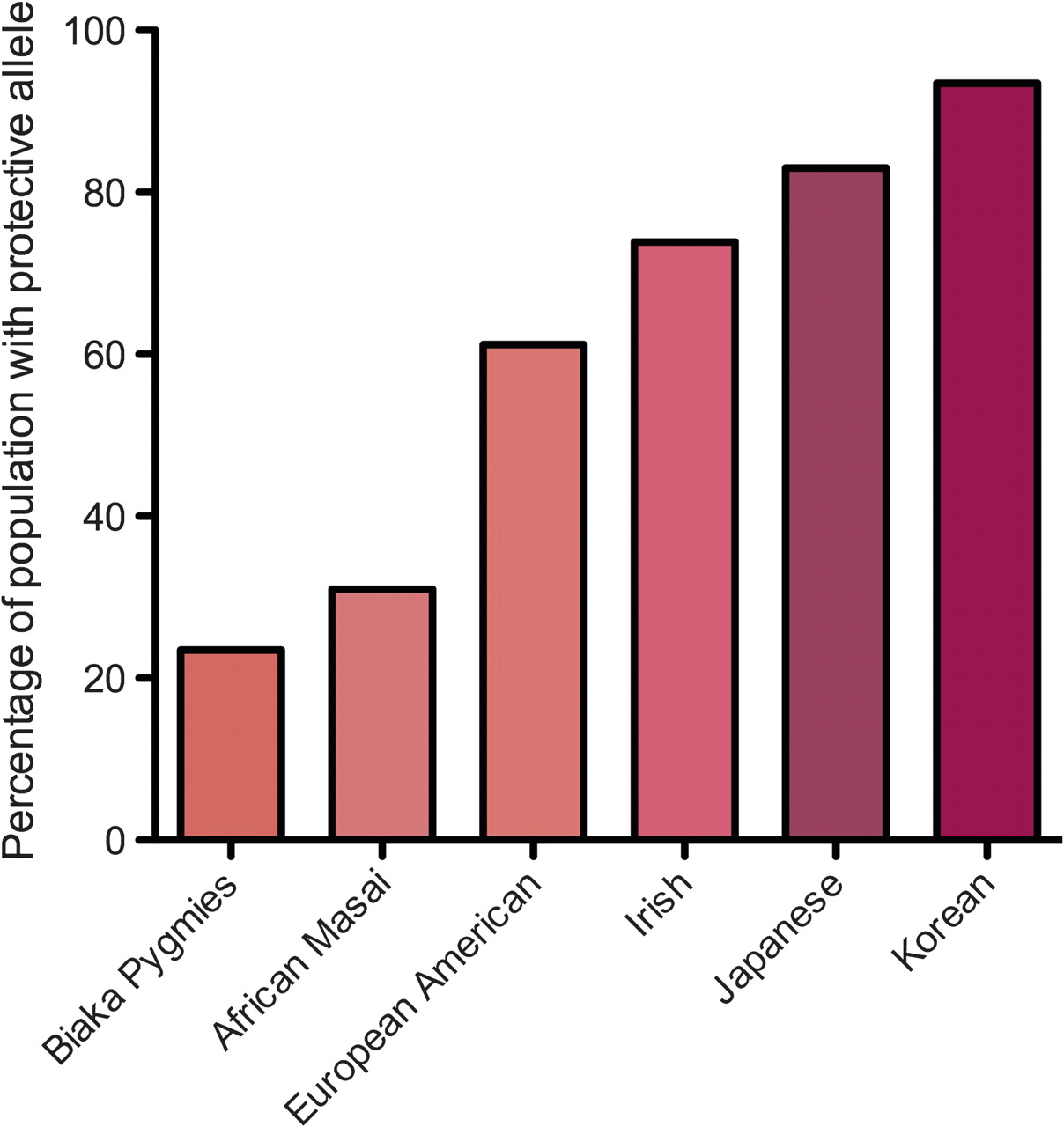
Figure 3 - Mean frequency of interferon λ3 (IFNλ3) protective allele in different ethnic groups. The protective allele is less common in African populations than in Asian populations, correlating with the known poor outcome from hepatitis C virus (HCV) infection in patients of African origin. Figure derived from data in Thomas et al.18
Shortly after the landmark publication by Ge et al,8 the same gene (using a candidate gene approach assessing SNP 12979860) was shown to play a key role in the spontaneous resolution of disease following primary infection in both African and Caucasian populations.18 This study showed that carriers of the risk allele were three times less likely to spontaneously clear the virus than homozygotes for the protective allele. This finding was subsequently supported in a large GWAS by Rauch et al9 in both HCV-infected patients and HIV and HCV co-infected patients. It is now quite clear that the IFNλ pathway plays a key role in natural host immunity as well as in the response to IFNα treatment of HCV infection.
Although the SNPs identified in the first phase of the GWAS above are linked to the IFNλ3 gene, they are not located within known regulatory elements or the coding region of the IFNλ3 gene itself. Rather, they have an expected association with ‘functional’ polymorphisms involved in the regulation or the function of the IFNλ3 gene product. This is because IFNλ3 has a distinct haplotype block (distinct regions of the host genome with SNPs that are in tight linkage disequilibrium with one another) that extends beyond the gene and the gene regulatory elements. The most significantly associated SNP identified by Ge and colleagues (rs12979860) lies 3 kb upstream of the IFNλ3 gene.8
In an attempt to identify the functional (or causal) polymorphism, these authors sequenced the IFNλ3 gene itself in 96 individuals and found two variants (one 37 base pairs upstream of the translation initiation codon (rs28416813) and a non-synonymous coding SNP (rs8103142) encoding an amino acid substitution within the IFNλ3 gene. However, these variants were so tightly linked that further genotyping of the entire cohort failed to show that any of these SNPs were uniquely linked to treatment outcome. Several studies have identified SNP rs8099917 as that most significantly associated with outcome.9–11 This SNP is located 8.9 kb from the end of transcription of IFNλ3 in the intergenic region between IFNλ2 and IFNλ3. Suppiah and colleagues further clarified the association and genotyped 20 SNPs in the IFNλ3, IFNλ2, IFNλ1 gene cluster region.10 Haplotype analysis identified a six-allele haplotype that was tagged by rs809917 (rs12980275, rs8105790, rs8103142, rs10853727, rs8109886 and rs8099917) linked to SVR. Notably, this haplotype encompasses regulatory elements from both IFNλ3 and IFNλ2. Tanaka and colleagues also sequenced 48 patients and established a similar seven-allele haplotype (rs8105790, rs11881222, rs8103142, rs28416813, rs4803219, rs8099917 and rs7248668) associated with SVR (OR 11.1 (p=1.35×10-25, 95% CI 6.6 to 18.6).11 Similarly, Rauch and colleagues, in searching for the causal variants, resequenced the IL28B locus in 47 individuals with and without the SNP rs8099917 and with different treatment outcomes and identified 21 SNPs, of which five tightly linked SNPs were thought to be potentially causative.9
Since multiple tightly-linked SNPs within and around the IFNλ3 gene are associated with SVR, it seems unlikely that sequence studies of the IFNλ3 region in association with known clinical outcome will identify which of these are causal. In vitro IFNλ3 functional assays combined with mutagenesis studies will determine the effect of these polymorphisms on IFNλ3 function, although even these may not prove causality.
A further note of caution: while it is generally assumed that common variants associated with disease in GWAS reflect the effect of a causal variant in close proximity, this is not necessarily so. Rare causal variants cannot be detected using GWAS technology, which only screens the human genome for common SNPs. An alternative possibility, therefore, is that multiple rare variants that lie distant from the common variant identified in GWAS occur stochastically and more frequently in association with the GWAS common variant. These associations have been termed ‘synthetic’.19 In this scenario a common variant identified in the GWAS will be erroneously linked to a disease due to its chance association with the true causal variants which lie undetected by GWAS methodology. This possibility has been demonstrated by the analysis of existing datasets showing that known rare causal mutations responsible for sickle cell disease and hearing loss can give rise to genome-wide significant synthetic associations that lie a considerable distance (2.5 Mb) from the causal mutations.20 These observations do not detract from the use of the GWAS variants in predicting treatment outcome in HCV infection, but they do raise the possibility that the biological mechanisms that underpin this are related to distant multiple rare genetic variants.
It has been known for some time that ethnicity affects treatment outcome of HCV infection, with viral clearance rates lowest in African-Americans, highest in East Asians and intermediate in European-Americans.5–7
We now know that the underlying rate of carriage of the IFNλ3 risk allele depends on the ethnic group of the population under study (figure 3). This suggests that ethnic differences in the IFNλ3 gene may explain, at least in part, the different response rates to therapy in different ethnic groups. Although one study did not find an association with IFNλ3 in the African-Americans studied, it is likely the group (n=53) was underpowered to identify the association.15
The data supporting a major role for the IFNλ3 gene in clinical outcome is less clear for genotypes 2 and 3 infection than for genotype 1. The largest study of 1362 patients by Rauch and colleagues9 and a smaller Spanish study14 found no association between genotypes 2 and 3 and IFNλ3 polymorphisms, although the treatment failure rate in both studies was very low (37/230 patients and 9/59 patients, respectively). In contrast, two other groups have found that IFNλ3 status can predict SVR in genotypes 2 and 3 infection, although in one of these studies the effect was restricted to patients who did not achieve a rapid virological response.13 15 Intriguingly, McCarthy and colleagues also showed that the protective IFNλ3 genotype is enriched in patients infected with genotype 3 HCV compared with genotype 1 (p=0.0007).15 The mechanism behind this observation is speculative but, since the protective allele is almost universal in Asian populations where genotype 3 infection has evolved,18 it seems plausible that this genotype may have evolved mechanisms to persist following acute infection in the face of the protective allele.
It remains to be seen how we can best use the information about IFNλ3 in clinical practice. In genotype 1 infection, IFNλ3 is the best predictor of treatment outcome when compared with other known baseline predictors including fibrosis score, viral load, gender and ethnicity.8 9 11 15 However, the sensitivity, specificity and predictive value of the IFNλ3 genetic variants currently identified are too low to be clinically useful alone. It has been calculated that the risk allele predicts non-response to treatment with a sensitivity of 57% and specificity of 63% with a positive predictive value of 64%.10 For homozygotes with the protective allele, SVR can be predicted with 65% sensitivity and 78% specificity in patients with genotype 1.15 It is plausible, however, that IFNλ3 alleic status will in future be incorporated into a treatment algorithm with other known predictors.
IFNλ3 and genetic technology
SNPs are single base pair mutations and are the main sites of genetic variation between individuals.
SNPs linked to the IFNλ3 gene predict both treatment response to PEG-IFNα/ribavirin and spontaneous clearance of HCV.
Individuals either heterozygous or homozygous for the risk allele are less likely to clear HCV infection than those homozygous for the protective allele.
African populations have a higher frequency of the risk allele than Caucasian or East Asian populations.
Carriers of the IFNλ3 risk allele are 2–12 times more likely to fail treatment than homozygotes for the protective allele.
The predictive value of the IFNλ3 alleles alone is insufficient to determine treatment outcome but may be a useful component of a treatment algorithm in the future.
The biology of interferon lambdas
The GWAS point to IFNλ as a key cytokine in the control of HCV infection. Is this biologically plausible? What do we know currently about the role and function of IFNλ?
Since their discovery in 2003,21 22 IFNλs (a type III IFN) have largely been considered a ‘poor relative’ of type I IFNs. Many functions appeared to overlap with type I IFNs and until now there has been little evidence of an independent and clinically significant role. Although the GWAS data suggest that IFNλ3 may be the type III IFN most relevant to the pathogenesis of HCV, historically the assessment of IFNλ biology has focused on IFNλ1 and IFNλ2.
Classification
IFNλs are classified within the class II cytokine family based on the similarity of their receptors. The class II family of cytokines consists of three types of IFNs (types I, II and III) as well as the interleukin 10 (IL-10)-related cytokines. Type I IFNs include IFNα and IFNβ, type II IFN is IFNγ and type III IFNs consist of IFNλ1, IFNλ2 and IFNλ3 which are also called IL29, IL28A and IL28B, respectively. IFNλ3 shares considerable amino acid sequence homology with IFNλ2 (96%) but less with IFNλ1 (81%).21 There are currently very limited available data comparing the different biological activity of the three different λ cytokines.
Although class II cytokines may be quite divergent at the amino acid level, they are structurally related with a shared α-helical pattern and signal through related heterodimeric transmembrane protein receptors. However, biological activity ultimately depends on the receptor cytoplasmic domains, which are not related, and these may trigger overlapping but different biological functions. This, in addition to the pattern of receptor distribution among different cell types, means that the different type II cytokines are functionally distinct.
Production of IFNλ
Broadly speaking, the IFNλ family is similar to IFNα in that it has both antiviral and immunomodulatory properties, but there are important differences. Whereas IFNα is produced by all nucleated cells, IFNλ may be produced by fewer cell types.23 Plasmacytoid monocyte-derived dendritic cells (pDC and MDDC) and macrophages produce IFNλ in response to influenza virus infection and/or to bacterial and viral molecular mimics (the Toll-like receptor agonists lipopolysaccharide and poly I:C).24 25 Interestingly, macrophage production of IFNλ is markedly enhanced by pre-culturing these cells with IFNα—evidence of interplay between the two pathways.26 27 There is evidence that IFNλ secretion may be influenced by cytokines—for example, pDCs produce an increased amount of IFNλ in response to IL-4 through a monocyte intermediate pathway.25 There is also evidence that IFNλ has a particular role in skin biology, being readily produced there by regulatory T cells and dendritic cells and acting upon keratinocytes and melanocytes.28 With particular relevance to HCV infection, hepatocyte tumour lines (HepG2 and HuH7.5),22 27 primary hepatocytes cultured ex vivo and liver tissue obtained from liver biopsy specimens produce IFNλ in response to viral infection.29 In contrast, primary human CNS tissue does not produce IFNλ.23
IFNλ receptor distribution and signalling
The IFNλ receptor also has a limited cellular distribution. IFNλs signal through a heterodimeric receptor composed of a short IL-10Rβ chain (also called IL-10R2) and a long chain IL-28Rα (also called IL-28R1).21 22 30 Whereas the short chain is ubiquitously expressed and is a receptor component of other type II-related cytokines (IL-10, IL-22, IL-26), the long chain is unique to IFNλ and has a limited tissue distribution.30 Hepatocytes from liver biopsy specimens have a high IFNλ receptor expression (IL-28Rα).23 30 31 However, this receptor is not found on fibroblasts, microvascular endothelial cells, adipocytes30 or primary CNS cells.23 30 It is likely that the limited receptor profile accounts for the relatively restricted side effect profile of IFNλ when administered to patients. Immune cells are clearly an important target of IFNs in general, however not all appear to express the IFNλ receptor. B cells have been shown to have the highest level of receptors, whereas the data on T cells are conflicting with both high and low receptor levels reported.30 31 Monocytes also appear to have a minimal receptor expression.30–32
All class II cytokines signal through the signal transducers and activators of transcription (STATs) and janus tyrosine kinases (JAK) and the biological properties of all IFNs—especially type I IFNs and the IFNλs—clearly overlap. In particular, both are produced in response to viral infection and have antiviral properties. However, within this signalling cascade there are subtle differences between the IFNs that are not fully understood. All class II cytokines appear to activate STAT1 and STAT3 and bind to IFNγ-activated sites (GAS) to various extents in the cell nucleus, resulting in the transcription of IFN-stimulated genes (ISGs). However, in addition, type I IFNs and IFNλ also activate IFN-stimulated response elements (ISRE) through the activation of STAT2 in association with STAT1 and IRF9 and which again results in the production of IFN-stimulated genes (figure 4).33 34
The signalling pathway of type III interferons (IFNs) compared with type II and type I IFNs. IFNλ binds the IFNλR1 chain leading to a conformational change in the receptor. It recruits a second short receptor chain (IL10R2). JAK 1 and Tyk 2 transphosphorylate the receptor chains and form phosphorylated tyrosine peptides on the IFNλR1 receptor chain. STAT proteins bind and are phosphorylated. They can then form homo- or heterodimers and migrate to the nucleus to bind gene regulatory elements (gamma-activated sequences, GAS). In the case of type I and type III IFNs, they can also bind interferon regulatory factor 9 (IRF9) in the cytoplasm and then migrate to the nucleus to bind interferon stimulated regulatory elements (ISREs) to regulate gene transcription.
Biological effects of IFNλ
Microarray experiments have shown that hundreds of ISGs are produced in response to IFN stimulation. The full spectrum of genes that are stimulated in response to different IFNs has not been defined, but it is clear that even different IFNs that bind to the same receptor may ultimately induce different genes35 36 and that mutations in a single amino acid in key locations in the IFNλ binding site can have a significant impact on antiviral activity, altering antiviral activity by up to 300-fold.37 Marcello et al evaluated ISG expression, protein production and HCV RNA replication using the HCV replicon system with cell culture HCV in response to IFNα and IFNλ stimulation.38 Both cytokines ultimately inhibited HCV replication in this system. However, the kinetics of STAT activation and the potential effector genes induced differed. In particular, IFNα genes peaked and declined rapidly whereas IFNλ genes increased steadily. Thus, the functional differences and the interplay between types I and III IFNs are likely to lie in the fine detail of this signalling pathway, yet to be fully elucidated for IFNλ, and in the differential distribution of cells that both produce IFNλ and are able to respond to IFNλ through the presence of appropriate receptors.
The effects of IFNλ, like that of IFNα, on immune cell function appear to be complex and diverse. This means that in vitro studies of discrete functions, such as the in vitro effects of IFNλ on an isolated cell type, may tell us little about the in vivo effects in a particular disease setting where a complex interplay of cell types and inflammatory stimuli is at work. With that proviso, IFNλ has been shown to decrease the production of Th2-type cytokines (IL-4, IL-5, IL-13, IL-14, IL-15), potentially favouring a Th1 immune pathway,39–41 increase T regulatory cells32 and augment CD8 T cell cytotoxicity and memory responses in macaques vaccinated with an HIV antigen.42 However, others have shown that immune cell subsets (monocytes, NK, T cells) are unresponsive to IFNλ1 and IFNλ2 and postulated that this is due to the production of a soluble receptor produced by peripheral blood mononuclear cells.30 32 43
The antiviral effects of IFNλ are clearer; in vitro IFNλ protects human cell lines against the cytopathic effects of vesicular stomatis virus (VSV) and encephalomyocarditis virus21 22 and, in hepatocyte models, IFNλ1 inhibits both HCV and HBV replication.31 38 44 However, some studies have shown that, unlike IFNα, IFNλ is unable to suppress HCV completely38 and all groups found it was less potent than IFNα. The antiviral effects of IFNλ on HCV infection in patients have also been demonstrated and are discussed further below.45 In this case, however, it is unclear if the antiviral effects are mediated directly or through the stimulation of immune cells, or both. It should be remembered that, in the case of chronic HCV infection, the presumed effects of IFNλ on viral clearance revealed in the GWAS are mediated through treatment with exogenous IFNα. How then might these cytokines interact with one another? Although IFNλ may be less potent than IFNα as a direct antiviral, together these cytokines may have an additive effect.27 38 46 In macrophages and hepatoma cell lines it has been shown that IFNα upregulates IFNλ1 production 1000-fold and IFNλ2/3 10–100-fold.26 27
Effect of IFNλ3 polymorphism on IFNλ biology
It is not yet clear how—or indeed if—IFNλ3 polymorphisms linked to the risk allele identified in the GWAS affect immune function or exert specific antiviral effects in HCV-infected patients. This will be a difficult task unless a causal IFNλ3 variant is identified, enabling the precise assessment of the relevant IFNλ3 variants in vitro. Currently, this assessment can only be made indirectly by observing immune function or HCV viral loads in people with and without the risk allele that lies a considerable distance from the IFNλ3 gene. It is currently not clear if IFNλ3 production by peripheral blood mononuclear cells (PBMCs) is associated with the IFNλ3-linked genetic variants; while two studies found that the protective allele is associated with increased levels of IFNλ3 in PBMCs,10 11 others have found it to be unrelated.8 However, a consistent finding appears to be that patients with the IFNλ3-linked protective allele have a higher pretreatment HCV viral load than those with the risk allele, with Ge and colleagues finding that those homozygous for the protective allele had a viral load of 6.35log10 IU/ml compared with 6.16 log10 IU/ml in those homozygous for the risk allele (p=1.21×10-10).8 15 This is paradoxical since a higher pretreatment viral load has been associated with a poor response to subsequent IFNα treatment. It should be noted, however, that the presence or absence of the risk allele does not associate with the clinically relevant viral load cut-off (5.78log10 IU/ml)47 48 that has been associated with response to IFNα therapy, and it is therefore likely that the effect of the protective allele on clinical outcome is independent of the effect of this allele on viral load.
IFNα and IFNλ ultimately exert their functions through the upregulation of ISGs. However, it has been repeatedly observed that a poor response to exogenous IFNα treatment is associated with a higher intrahepatic ISG expression before treatment.49 50 One plausible explanation is that, in this setting, the IFNα pathway (and thus ISG expression) is already maximally stimulated and is therefore unresponsive to further exogenous IFNα treatment. A recent study has shown that the protective allele identified in the GWAS (rs8099917) does not correlate with intrahepatic IFNλ3 production.49 Technically there are reasons why this might be so; for example, the low levels of IFNλ3 detected in general make correlations difficult and by the fact that, owing to sequence homology between IFNλ3 and IFNλ2, quantification by real-time PCR does not distinguish between these cytokines. However, the protective allele does correlate with a lower expression of ISGs and, in those with the risk allele, lower levels of intrahepatic IFNλ3 were associated with higher ISG (Mx1 IFI44 and IFIT1) expression.49 In fact, the assessment of ISGs in response to IFNs is not straightforward given that many hundreds of these are expressed in liver tissue that is composed of hepatocytes and also of immune and other cell types. For example, in the study by Honda and colleagues, 1359 genes were differentially expressed between individuals with and without the protective allele. Pathway analysis suggested that IFN action and apoptosis were upregulated in the liver of those with the risk allele, whereas immune cell (B, NK and dendritic cells) genes were expressed in those with the protective allele.49
This is supported by the observation that ISG expression is upregulated in Kupffer cells (liver macrophages) of IFNα responders compared with non-responders, whereas ISGs are upregulated in hepatocytes of non-responders compared with responders.51 In other words, the protective effect of the IFNλ3 allele may relate to the effects on or by ‘visiting’ cell populations rather than hepatocytes themselves.
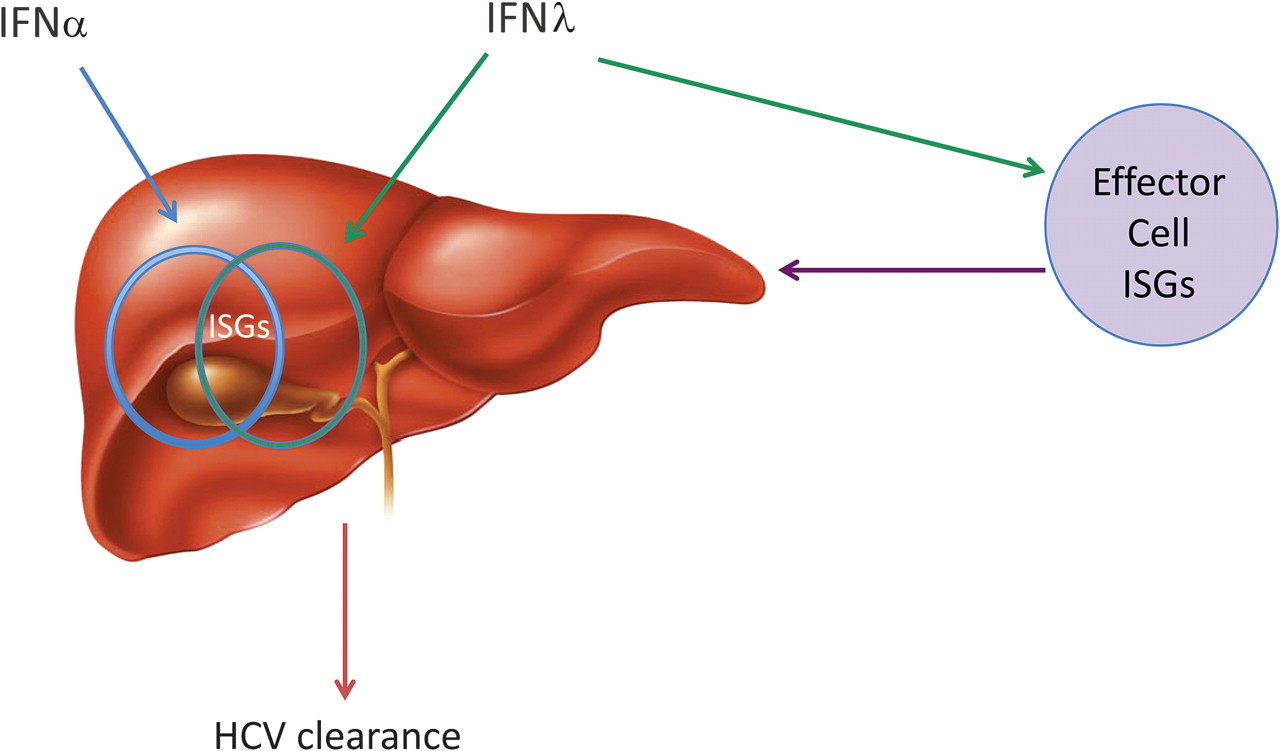
In the setting of acute HCV infection, assuming the GWAS have correctly identified a genetic signal linked to IFNλ3, protective IFNλ3 variants could mediate viral clearance through immune stimulation or direct antiviral effects through the stimulation of the ‘right’ ISGs. Exactly what these ISGs are and in which cells they are produced is not yet clear. In the setting of chronic infection where IFNλ3 variants also appear to be associated with response to exogenous IFNα, the protective IFNλ3 variant may well be linked to a complex intrahepatic ISG profile consisting of the ‘right’ ISGs produced by particular cell types (figure 5). Undoubtedly, identifying exactly what the right ISGs are is a challenging but key question that will be addressed in months to come.
{kind=link}
{kind=link}

Figure 5
Model of the possible action of interferon λ (IFNλ) in chronic hepatitis C virus (HCV) infection. In chronic HCV infection, IFNα upregulates hepatic interferon-stimulated genes (ISGs) (represented by blue circle) but this fails to clear HCV. Addition of IFNλ may result in stimulation of different ISGs (represented by green circle) and therefore HCV clearance. The IFNλ polymorphism could impact on the expression of the right hepatic ISGs directly or through the stimulation of effector cell ISGs resulting in HCV clearance.
The biology of IFN lambdas
IFNλ is a type III IFN of which there are three types: IFNλ1, IFNλ2, IFNλ3 (IL29, IL28A, IL28B).
Plasmacytoid dendritic cells are the main producers of IFNλ stimulated by viruses and viral or bacterial mimics.
IFNλ has a restricted receptor distribution and therefore restricted targets.
INFλ has antiviral, antitumour and immunomodulatory effects.
Upregulated hepatic ISGs pretreatment are associated with the IFNλ3 risk allele and with poor treatment outcome.
The effect of IFNλ3 on HCV clinical outcome is likely to be a complex interplay between IFNα, the virus itself, ISGs and immunomodulation.
Clinical studies
IFNλ is potentially an attractive alternative to IFNα for the treatment of HCV infection. IFNα has been used to treat HCV since the discovery of the virus two decades ago. Over this time, important modifications to IFNα—such as the addition of polyethylene glycol (PEG) to increase drug half-life and the addition of ribavirin to treatment regimes—have been incorporated. Although in the coming years HCV protease and polymerase therapies will add to the anti-HCV drug armoury, the rapid emergence of HCV variants which are resistant to these new drugs means that IFNα will probably remain an essential component of treatment regimens. However, treatment with IFNα is associated with a very significant adverse event profile. Thrombocytopenia, neutropenia and psychiatric complications such as anxiety, depression and sleep disturbance are particularly troubling. An IFN with potent antiviral effects and a more favourable side effect profile would revolutionise the treatment of HCV. In vitro experiments have shown that that IFNλ has a limited effect on CNS and haematopoietic cells. So, with its promising antiviral activity in the liver but restricted receptor distribution, IFNλ seems like the ideal candidate for development into a therapeutic agent.
Since the biological potential of IFNλs in general was recognised before the studies that implicated IFNλ3 in particular, clinical trials to date have not focused on IFNλ3 but rather on IFNλ1. Zymogenetics (in collaboration with Bristol-Myers Squibb) are assessing PEG-IFNλ1 in clinical studies which, while showing promising antiviral activity, have also uncovered a different but nevertheless significant side effect profile from that seen with IFNα. A phase I dose escalation study of PEG-IFNλ1 was presented in 2007 as an abstract
(http://www.zymogenetics.com/products/documents/HEP_DART_PEG_IFN_12-07.pdf).
Volunteers received a single subcutaneous injection of PEG-IFNλ1. The study was stopped after volunteers received 7.5 μg/kg, below the planned target maximum of 40 μg/kg, after four volunteers developed significantly elevated transaminases. As there was evidence of bioactivity at lower doses that were not associated with hepatotoxicity, and since peak serum concentrations at tolerated doses were higher than those shown to have antiviral effect in vitro,31 38 a phase I study in HCV-infected patients followed.
In this 4-week open-label trial, patients with genotype-1 HCV infection received Peg-IFNλ1 weekly or two-weekly.45 The study included both treatment-naïve patients (n=7) and patients with previous virological relapse with standard therapy (n=49). Despite the lower doses in this trial (0.5–3 μg/kg), six patients stopped or had treatment withheld due to elevated transaminase levels and four patients had raised lipase or amylase elevations which resolved after stopping PEG-IFNλ1. Although adverse effects were more common in the highest dose group, it is noteworthy that, at only 1.5 μg/kg, one patient developed severe hepatotoxicity with alanine transaminase levels of 667 IU/ml and bilirubin levels of 348.9 μmol/l. Furthermore, one patient developed idiopathic thrombocytopenic purpura and three patients developed antibodies to IFNλ1 during this short treatment course. Two patients previously treated with IFNα had cross-reactive antibodies to IFNλ at baseline (although only one of these were neutralising antibodies), suggesting that previous treatment with IFNα may limit IFNλ efficacy in some cases.
In keeping with the in vitro data showing a limited IFNλ receptor distribution,23 30 there was a noticeable decrease in IFNα-type side effects. Fatigue was the most common side effect (28.6%) followed by nausea and myalgia (12.5% and 10.7%, respectively). However, insomnia and influenza-like illness were only experienced in 5.4%, well below rates reported with IFNα and significant neutropenia did not occur. This trial also showed that IFNλ1 is capable of antiviral activity in vivo. In patients receiving a dose of ≥1.5 μg/kg/week, 23/24 patients with treatment relapse achieved a fall in viral load of ≥2log10 and four of these achieved a rapid virological response, defined as undetectable viraemia at 4 weeks. Interestingly, all eight African-American patients (a group known to respond poorly to IFNα treatment) in this study had a decline in viral load of >2log10 at the end of treatment. The association of virological response to PEG-IFNλ and IFNλ genotype has very recently been assessed in an ongoing phase II study (published in abstract form only) of PEG-IFNλ1 or PEG-IFNα with ribavirin combination therapy for 24–48 weeks, with a 2-week lead-in period of PEG-IFNλ1 or PEG-IFNα alone, in patients with genotypes 1–4 HCV infection.52 Interim results from patients receiving 120–240 μg PEG-IFNλ1, revealed a 71% viral response (VR) (≥2log10 reduction in viral load) at week 4 in those with a favourable genotype compared with 25% VR in those carrying the risk allele.52 In this study, grade 2/3 hepatotoxicity was again observed (alanine transaminase or aspartate aminotransferase 2.5–20 times upper limited of normal) in 20% of patients on PEG-IFNλ1 which was, however, successfully managed with dose reduction.52
IFNλ may also in the future be considered both in the context of cancer therapy since this cytokine has been shown to suppress tumorigenesis in vitro27 31 38 and also as an immunomodulatory agent in the suppression of Th2-associated diseases such as allergy and atopy.39–41 IFNλ may have a particular role in asthma since it suppresses IL-13 (known to be elevated in patients with asthma),39 and deficient IFNλ production in the bronchi of patients with asthma is associated with an increase in respiratory infections in this population.53
The real utility of PEG-IFNλ1 in the treatment of HCV will become more apparent following the final results from the ongoing phase II trial described above which will assess both safety and efficacy.52 Ultimately, new agents for HCV will need to be tolerable in the setting of pre-existing liver inflammation and poor hepatic reserve associated with liver cirrhosis.
Clinical studies
A phase I trial of PEG-IFNλ1 has shown promising antiviral activity in both treatment relapsers and treatment-naïve patients with HCV.
Hepatotoxicity has been associated with the clinical use of PEG-IFNλ1.
Impaired IFNλ production may lead to increased susceptibility to viral infections.
IFNλ3 provides the opportunity to apply pharmacogenomics to HCV treatment.
Conclusion
The future is likely to bring IFNλ3 and this exciting cytokine family further into the limelight, with head-to-head clinical trials between PEG-IFNλ1 and PEG-IFNα underway for both HCV and HBV. Importantly, although genetic studies have now suggested that IFNλ3 has a pivotal role in HCV clinical outcome, most work to date has been with IFNλ1. IFNλ3 may yet be developed as an alternative HCV treatment, particularly in those with the risk polymorphism, and genotyping for IFNλ3-related SNPs may become part of a treatment decision algorithm. The differential effect of IFNλ treatment on patients with and without the IL28B polymorphism is clearly an area worth further evaluation. Pharmacogenomic treatment approaches are one of the future challenges of medicine, and HCV provides the ideal opportunity to tailor potentially toxic therapies to individuals most likely to benefit. Whether work on this cytokine is applicable to genotypes other than 1 is still an open question, as is establishing the ideal therapeutic window for treatment with type III IFNs which have demonstrated hepatotoxicity in both healthy volunteers and patients with HCV infection.
Furthermore, elucidating the pathway by which the IFNλ3 polymorphism affects the response to acute infection and to IFN therapies remains an important future challenge. It is likely that defining the pattern of gene stimulation by IFNλ in different cell types, as well as potential structural differences in IFNλ3, will be critical in solving this puzzle. In particular, investigating the immunomodulatory effect of IFNλ may well be key to unlocking the mechanism behind both its antiviral effect and its toxicities. Overall, the discovery of the link between the IFNλ3 locus and HCV disease outcome has been a huge step forward for the field. However, while it has provided momentum for clinicians and basic scientists alike, the actual route ahead still needs careful charting. Nevertheless, while the destination is not yet clear, the journey itself will certainly be interesting.
Footnotes
Funding CK is funded by the Oxford NIHR Biomedical Research Centre and Wellcome Trust UK, PK is funded by the Wellcome Trust UK and the Oxford NIHR Biomedical Research Centre, EB is funded by the Medical Research Council UK and the Oxford NIHR Biomedical Research Centre, PK is funded by the James Martin School for 21st Century and NIH V19AI082630.
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license.
See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode
Full text
No comments:
Post a Comment