Safety and on-treatment efficacy of telaprevir: the early access programme for patients with advanced hepatitis CM Colombo1, I Fernández2, Abdurakhmanov3, A Ferreira4, I Strasser5, P Urbanek6, C Moreno7, A Streinu-Cercel8, A Verheyen9, W Iraqi10, R DeMasi11, A Hill12, J M Läuffer13, I Lonjon-Domanec10, H Wedemeyer14
+ Author Affiliations
1Department of Medicine, Division of Gastroenterology, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Università degli Studi di Milano, Milan, Italy
2Hospital Universitario 12 de Octubre, Sección de Aparato Digestivo, Madrid, Spain
3I.M. Sechenov First Moscow State Medical University, E. M. Tareev Clinic for Nephrology, Internal and Occupational Medicine, Moscow, Russia
4Viral Hepatitis Division of Infectious Disease, Outpatient Clinic to HIV, Federal University of São Paulo, São Paulo, Brazil
5AW Morrow Gastroenterology and Liver Centre, Royal Prince Alfred Hospital, University of Sydney, Sydney, Australia
6Department of Internal Medicine, First Medical Faculty, Charles University, and Central Military Hospital Prague, Prague, Czech Republic
7Liver Unit, Department of Gastroenterology, Hepatopancreatology and Digestive Oncology, Erasme University Hospital, Université Libre de Bruxelles, Brussels, Belgium
8Carol Davila University of Medicine and Pharmacy, National Institute for Infectious Diseases, "Prof. Dr. Matei Bals", Bucharest, Romania
9Janssen Pharmaceutica, Beerse, Belgium
10Janssen Pharmaceuticals, Paris, France
11Janssen Research and Development, Titusville, New Jersey, USA
12Janssen Research and Development, High Wycombe, UK
13Janssen-Cilag AG, Zug, Switzerland
14Medizinische Hochschule Hannover, Hannover, Germany
Correspondence to Professor Massimo Colombo, 1st Division of Gastroenterology, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Via F. Sforza 35, Milan 20122, Italy; massimo.colombo@unimi.it
Received 15 July 2013
Revised 3 October 2013
Accepted 15 October 2013
Published Online First 7 November 2013
Abstract
Background and aim
Severe adverse events (AEs) compromise the outcome of direct antiviral agent-based treatment in patients with advanced liver fibrosis due to HCV infection. HEP3002 is an ongoing multinational programme to evaluate safety and efficacy of telaprevir (TVR) plus pegylated-interferon-α (PEG-IFNα) and ribavirin (RBV) in patients with advanced liver fibrosis caused by HCV genotype 1 (HCV-1).
Methods
1782 patients with HCV-1 and bridging fibrosis or compensated cirrhosis were prospectively recruited from 16 countries worldwide, and treated with 12 weeks of TVR plus PEG-IFN/RBV, followed by 12 or 36 weeks of PEG-IFN and RBV (PR) alone dependent on virological response to treatment and previous response type.
Results
1587 patients completed 12 weeks of triple therapy and 4 weeks of PR tail (53% cirrhosis, 22% HCV-1a). By week 12, HCV RNA was undetectable in 85% of naives, 88% of relapsers, 80% of partial responders and 72% of null responders. Overall, 931 patients (59%) developed grade 1–4 anaemia (grade 3/4 in 31%), 630 (40%) dose reduced RBV, 332 (21%) received erythropoietin and 157 (10%) were transfused. Age and female gender were the strongest predictors of anaemia. 64 patients (4%) developed a grade 3/4 rash. Discontinuation of TVR due to AEs was necessary in 193 patients (12%). Seven patients died (0.4%, six had cirrhosis).
Conclusions
In compensated patients with advanced fibrosis due to HCV-1, triple therapy with TVR led to satisfactory rates of safety, tolerability and on-treatment virological response with adequate managements of AEs.
Significance of this studyWhat is already known on this subject?The treatment outcome for patients with chronic hepatitis C due to genotype 1 of HCV has remarkably improved following the addition of the oral HCV protease inhibitors boceprevir (BOC) or telaprevir (TVR) to pegylated interferon+ribavirin (RBV) (PR) therapy.
However, advanced hepatic fibrosis may limit both the access as well as the response rates of patients to triple therapy with significant issues of tolerability and safety.
A French observational study with TVR- or BOC-based triple therapy in patients with cirrhosis reported high frequency of anaemia and severe adverse events (AEs) particularly in patients with low platelet counts and low serum values of albumin (beyond the selection criteria of registration trials).
What are the new findings?We gained further insights into the safety and efficacy of TVR+PR treatment in 1587 patients with advanced liver fibrosis or cirrhosis due to HCV-1 who were enrolled in an open label expanded access programme to TVR-based regimen and were selected following registration trials criteria for platelets, neutrophils and albumin levels.
During 16 weeks of treatment (12 weeks of triple therapy followed by 4 weeks of PR) anaemia was the main side effect as 59% of patients developed grade 1–4 anaemia (grade 3/4 in 31%), 630 (40%) dose reduced RBV, 332 (21%) received erythropoietin and 157 (10%) were transfused.
Treatment was safe as 12% of patients had to discontinue TVR due to AEs and only seven died (0.4%, six with cirrhosis).
At the end of triple therapy serum HCV RNA was undetectable in the vast majority of previous untreated patients and importantly in 72% of 436 patients who had had a prior null response to PR. The latter is the most difficult to cure population that was under-represented in a previous field practice study in France.
How might it impact on clinical practice in the foreseeable future?In compensated patients with advanced liver fibrosis due to HCV-1 who fulfilled the selection criteria of registration trials, 16 weeks of TVR triple therapy proved to be safe, tolerated and effective.
IntroductionChronic infection with HCV is a leading cause of liver-related morbidity and mortality worldwide, for which an effective antiviral treatment with pegylated-interferon (PEG-IFN) and ribavirin (PR) has been available since early 2000.
1 ,
2 The treatment outcome for difficult to cure patients, such as those chronically infected with HCV genotype 1 (HCV-1), has remarkably improved following the addition of the oral HCV protease inhibitors (PI) boceprevir (BOC) or telaprevir (TVR) to PR therapy.
3–5 While eradication of HCV by PI+PR therapy is expected to greatly extend treatment success rates, resulting in a reduced risk of liver-related mortality,
6–8 factors that may limit both the access as well as the response rates of patients to triple therapy have been identified. Perhaps the most relevant is the presence of advanced hepatic fibrosis,
9–11 which affects tolerability and safety of triple therapy. The impact of this factor on treatment access and response rates is driven by an increased rate of myelosuppression-related adverse events (AEs) such as anaemia and infections in patients treated with triple therapy.
10–13 In addition, suboptimal dosing due to poor treatment tolerability may impact the outcome of PI+PR therapy in patients with advanced fibrosis, leading to reduced rates of virus eradication and also to unaffordable cost to utility ratios.
14 A French observational study with TVR- and BOC-based triple therapy in patients with cirrhosis revealed that 33%–46% of the patients had been ineligible for registration studies with one of these PIs due to their severe level of liver disease. Up to 11.7% of patients had to discontinue therapy until week 16 due to the onset of AEs including anaemia, neutropenia, rash, clinical decompensation and bacterial infections. In addition, more than half of the patients on TVR required treatment with bone marrow stimulating factors and 16.1% received blood transfusions to manage treatment-related anaemia.
15 Another real-world study in Germany showed a high frequency of anaemia and SAEs in patients with cirrhosis who were treated with triple therapy, particularly in patients with low platelet counts.
16 Therefore, the safety of triple therapy is now being perceived by hepatologists as a potential barrier to its use in patients with the most need, such as those with advanced fibrosis, who are more often vulnerable to myelosuppression-related complications.
To gain further insights into the safety and efficacy of TVR+PR treatment in patients with advanced liver fibrosis or cirrhosis due to HCV-1 infection, an open label expanded access programme (EAP) was launched in late 2011 involving 16 nations worldwide. Cumulatively, the programme enrolled more than 2000 treatment naive or experienced patients with bridging fibrosis or cirrhosis. We report here the analysis of the on-treatment virological response and safety in the first 1587 patients who reached week 16 of therapy.
Patients and methods
PatientsFrom August 2011 to March 2013, compensated (Child–Pugh A) patients with bridging fibrosis or cirrhosis due to HCV-1 infection were enrolled in the TVR EAP at study centres located in 16 countries across Europe, South America and Australasia. Key inclusion criteria were male and female gender; ages 18–70; infection with HCV-1; quantifiable serum HCV RNA; and documentation of liver fibrosis assessed by liver biopsy or a non-invasive test like Fibrotest or Fibroscan showing severe fibrosis (Metavir F3 or Ishak 3–4 (S3,4)) or cirrhosis (Metavir F4 or Ishak 5–6 (S5,6)). Eligible patients had to have an absolute neutrophil count >1500/mm3, a platelet count >90 000/mm3 and haemoglobin (Hb) >12 g/dL for women and 13 g/dL for men. Patients were excluded if they had history or other evidence of decompensated liver disease, hepatocellular carcinoma, or if they were infected or co-infected with HCV other than genotype 1, HBV, or HIV or had a history of alcohol abuse. Additionally, patients were excluded if they had a history of receiving HCV protease or polymerase inhibitors. The enrolment criteria for the TVR EAP were similar to the REALIZE study.
12The protocol was signed by the principal site investigators and approved by the independent ethics committee at each participating study centre. All patients provided written informed consent prior to the conduct of any procedures for the EAP.
TreatmentDuring the first 12 weeks of the programme, all patients received oral administration of TVR at a dose of 750 mg every 8 h in combination with PR. The type (α 2a vs α 2b PEG-IFN; copegus vs rebetol) and doses of PR were selected according to local guidelines. After week 12, PR was administered for another 12 or 36 weeks depending on the virological response to treatment and/or previous response type. The concomitant administration of PR followed label recommendations.
17 In patients with bridging fibrosis who were treatment naive or prior treatment relapsers, PR was administered for another 12 or 36 weeks based on virological response to treatment as measured by week 4 and 12 plasma HCV RNA levels. Patients with undetectable HCV RNA at weeks 4 and 12 received an additional 12 weeks of PR alone (total treatment duration of 24 weeks); patients with detectable HCV RNA (but not meeting stopping rules) at either week 4 or 12 received an additional 36 weeks of PR alone (total treatment duration of 48 weeks). Patients with bridging fibrosis with prior partial or null response to previous treatment with PR including a viral breakthrough and all patients with cirrhosis were treated for a subsequent 36 weeks. At weeks 4 and 12, patients were assessed as to whether they met the predefined stopping rules based on virological response. If at either time point the HCV RNA levels were greater than 1000 IU/mL, all treatment was permanently discontinued.
Measures of disease severity and treatment efficacyLiver fibrosis was staged on histological specimens obtained by percutaneous liver biopsy using either the Metavir or Ishak score or by Fibrotest or Fibroscan. Fibroscan cut-off to diagnose bridging fibrosis and cirrhosis was ≥9.5 kPa and ≥12.5 kPa, respectively.
18 The Child–Pugh score system was used to define the clinical status of patients, Child–Pugh A stage indicating compensated liver disease.
A range of assays were used to measure HCV RNA levels at local investigational sites. The majority of sites used the Roche COBAS TaqMan or Abbott RealTime assays (96%). Roche COBAS TaqMan (versions 1 and 2) has a lower limit of quantification (LLOQ) of 15–25 IU/mL depending on serum volume and the method of RNA extraction with a lower limit of detection (LLOD) of 10 IU/mL. Abbott RealTime has an LLOQ of 12 IU/mL and an LLOD of 10–12 IU/mL.
In the intent to treat analysis, the number of patients who had HCV RNA levels below LLOD was calculated for the overall population (n=1587) and in subgroups by prior treatment history.
Safety assessmentsThroughout the treatment period and follow-up, safety assessments were carried out including laboratory assessments, physical examinations, evaluation of vital signs and the reporting of AEs. Any clinically significant abnormalities persisting at the end of the EAP/early withdrawal were followed by the investigator until a resolution or clinical stable endpoint was reached.
The DAIDS criteria were used to grade AEs
19 except for rash which had protocol-specific definitions of severity grades: grade 1 mild, localised to one or several isolated sites; grade 2 moderate, diffuse skin eruption involving up to 50% of the body surface area; grade 3 severe, involving more than 50% of the body surface area or with significant systemic signs or symptoms; and grade 4 life-threatening, diagnosis of generalised bullous eruption, Steven Johnson syndrome or TEN. For patients who experienced grade 1 or 2 rash, medical management was left to the discretion of the investigator. For patients whose grade 2 rash progressed to grade 3, and for those experiencing grade 3 rash, TVR was permanently discontinued. If the rash did not improve, symptomatically or objectively within 7 days following TVR discontinuation, R was also discontinued. Immediate and permanent discontinuation of all study drugs was mandatory for all patients diagnosed with grade 4 rash.
Hb levels were assessed before treatment and at weeks 2, 4, 8 and 12, and as clinically appropriate thereafter; additional visits could be performed at the discretion of the investigator. Anaemia was defined as an AE by the investigator in the case report form, according to the following guidelines: grade 1 Hb values between 10.0 and 10.9 g/dL or any decrease from baseline between 2.5 and 3.4 g/dL; grade 2 Hb values between 9.0 and 9.9 g/dL or any decrease of Hb between 3.5 and 4.4 g/dL; grade 3 Hb values between 7.0 and 8.9 g/dL or any decrease of Hb ≥4.5 g/dL; and grade 4 an Hb value less than 7.0 g/dL. If anaemia developed during treatment, R dose was modified according to label recommendations. TVR was discontinued only if reductions of R dose or discontinuation did not result in an improvement of anaemia. TVR dose reductions were prohibited and TVR could not be reinitiated if treatment was discontinued. Use of blood transfusions, erythropoietin (EPO) or iron-based products were allowed during the trial.
Statistical methodsFisher's exact tests were used to detect differences in the prevalence of AEs between patients who were F3 versus F4 at baseline. Multivariate linear regression was used to identify baseline factors associated with a higher probability of extended rapid virological response (eRVR) (defined as HCV RNA undetectable at both weeks 4 and 12) and development of anaemia (defined as Hb below 10 g/dL at any time on treatment).
Results
Baseline characteristicsPatient demographics and disease severity are shown in
table 1 A,B. A total of 746 patients (47%) had bridging fibrosis and 835 patients (53%) a cirrhosis (F4 or S5,6).
View this table:
In this window In a new window
Table 1
Baseline characteristics of the study patients according to disease severity*
Disease stage was assessed by Fibroscan in 1149 patients (72%), biopsy in 308 patients (19%) and fibrosis markers in 130 patients (8%). Fibrosis stage was classified by Metavir in 1412 patients (89%) and Ishak in 175 patients (11%). The mean and SD of the baseline score of Fibroscan was 11.6 kPa (2.8) for patients classified as F3 at baseline, and 25.0 kPa (12.6) for patients classified as F4 at baseline.
Overall, the mean age of the patients was 53; 1012 patients (64%) were male and 1557 (98%) were white. In all, 357 patients (22%) were infected with HCV-1a and 1055 (66%) had HCV RNA greater than or equal to 800 000 IU/mL. At baseline, 374 patients (24%) had grade 1–3 thrombocytopenia (platelets <125 000/mm3), while 134 patients (8%) had a grade 2–4 thrombocytopenia (platelets <100 000/mm3). A total of 23 patients (1%) had grade 1–3 reductions in serum albumin (<3.5 g/dL).
Among the 532 out of 835 cirrhotic patients with currently available information, 78 patients (14.7%) had either grade or presence of oesophageal varices reported.
Overall, 321 patients (20%) were treatment naive, 531 (33%) were prior treatment relapsers, 203 (13%) were prior partial responders, 436 (27%) were prior null responders and 49 (3%) had had a viral breakthrough. In all, 47 patients (3%) had had an unspecified previous non-response. The demographic and clinical characteristics were similar between the bridging fibrosis and cirrhosis study groups.
Efficacy
Overall, 82% of patients at week 4 had a serum HCV RNA level less than 25 IU/mL, a rate that increased to 86% at week 12. Furthermore, 60% and 82% of patients had undetectable HCV RNA at weeks 4 and 12, respectively. The percentage of treatment naive patients who had undetectable HCV RNA levels at week 12 was 85% whereas among treatment experienced patients, 88% of treatment relapsers, 80% of partial responders, 72% of null responders and 84% of viral breakthrough patients had undetectable HCV RNA levels at week 12.
Figure 1 shows the outcome of treatment at weeks 4 and 12 for each subgroup of patients with respect to previous PR treatment. At week 12, null responders had a significantly higher rate of virological failure (14%) than the other groups (treatment naives (4%), prior relapsers (2%) and partial responders (5%)). In all, 37 patients (2%) stopped TVR at week 4 having met the futility rules criteria. Three patients (0.2%) continued triple therapy despite having met the week 4 virological criteria for anticipated TVR interruption. The number of patients discontinuing triple therapy for AEs was similar in all groups (7% for treatment naive, 4% for prior relapsers, 7% for partial responders and 7% for null responders).

Figure 1
Outcome of treatment at weeks 4 and 12, by prior treatment. Shown are the week 4 and 12 outcomes of treatment by subgroup: treatment naive (n=321), prior treatment relapsers (n=531), previous partial responders (n=436) and previous null (n=203). Data for patients who had previously experienced viral breakthrough (n=49) and whose prior response to treatment was unspecified (n=47) have not been represented.
In multivariate analysis, four baseline factors were associated with a higher chance of eRVR: baseline viral load <800 000 IU/mL (OR=1.47, 95% CI 1.18 to 1.85), genotype 1b (OR=1.52, 95% CI 1.16 to 1.96), α-fetoprotein <10 pg/mL (OR=2.36, 95% CI 1.82 to 3.23) and naive, relapser or prior partial response versus prior null response (OR=2.0, 95% CI 1.56 to 2.5). The rates of eRVR were 20/50 (40%), 77/235 (33%), 262/500 (52%), 349/584 (60%) and 173/200 (79%) for patients with 0, 1, 2, 3 or 4 of these predictive factors, respectively. There was no association between eRVR and the type of PEG-IFN used.
Safety and tolerabilityThrough week 16, 1014 (64%) patients experienced grade 2–4 AEs that were considered related to TVR treatment. The most common AEs were anaemia (n=698, 44%), rash (n=201, 13%), thrombocytopenia (n=120, 8%), pruritus (n=95, 6%) and asthenia (n=91, 6%). Patients with cirrhosis developed more AEs than those with bridging fibrosis (67% vs 60%, p=0.01). Overall, 12% of patients experienced AEs that ultimately led to TVR discontinuation (
table 2). Of the 1587 patients, seven patients died during the PR tail as a consequence of hepatic failure, pneumonia, haemorrhage, septic shock or ischaemic colitis leading to subsequent multi-organ failure. Detailed results are shown in
table 3.
Table 2
Reasons for discontinuation of telaprevir and the incidence of the most common grade 2–4 drug-related AEs and serious AEs
Table 3
Adverse events with fatal outcome (n=1587)
By week 16, 931 patients (59%) developed any grade anaemia (
table 4). Among patients with bridging fibrosis and cirrhosis, 55% and 62%, respectively, had any grade anaemia, with 25% and 29% of the patients developing grade 3 anaemia and 3% and 4% developing grade 4 anaemia, respectively. As expected, grade 2–4 anaemia judged to be related to TVR treatment occurred more frequently in patients with cirrhosis than in those with bridging fibrosis (41% vs 47%, p<0.01,
table 2). For treatment of anaemia, 630 patients (40%) underwent R dose reduction, 332 (21%) received EPO and 157 (10%) received a blood transfusion. There was combined use of EPO and blood transfusion in 74 patients (5%), EPO and R dose reduction in 234 patients (15%) and combined use of transfusion and R dose reduction in 141 patients (9%).
View this table:
Table 4
Week 16: Prevalence and management of anaemia by fibrosis stage
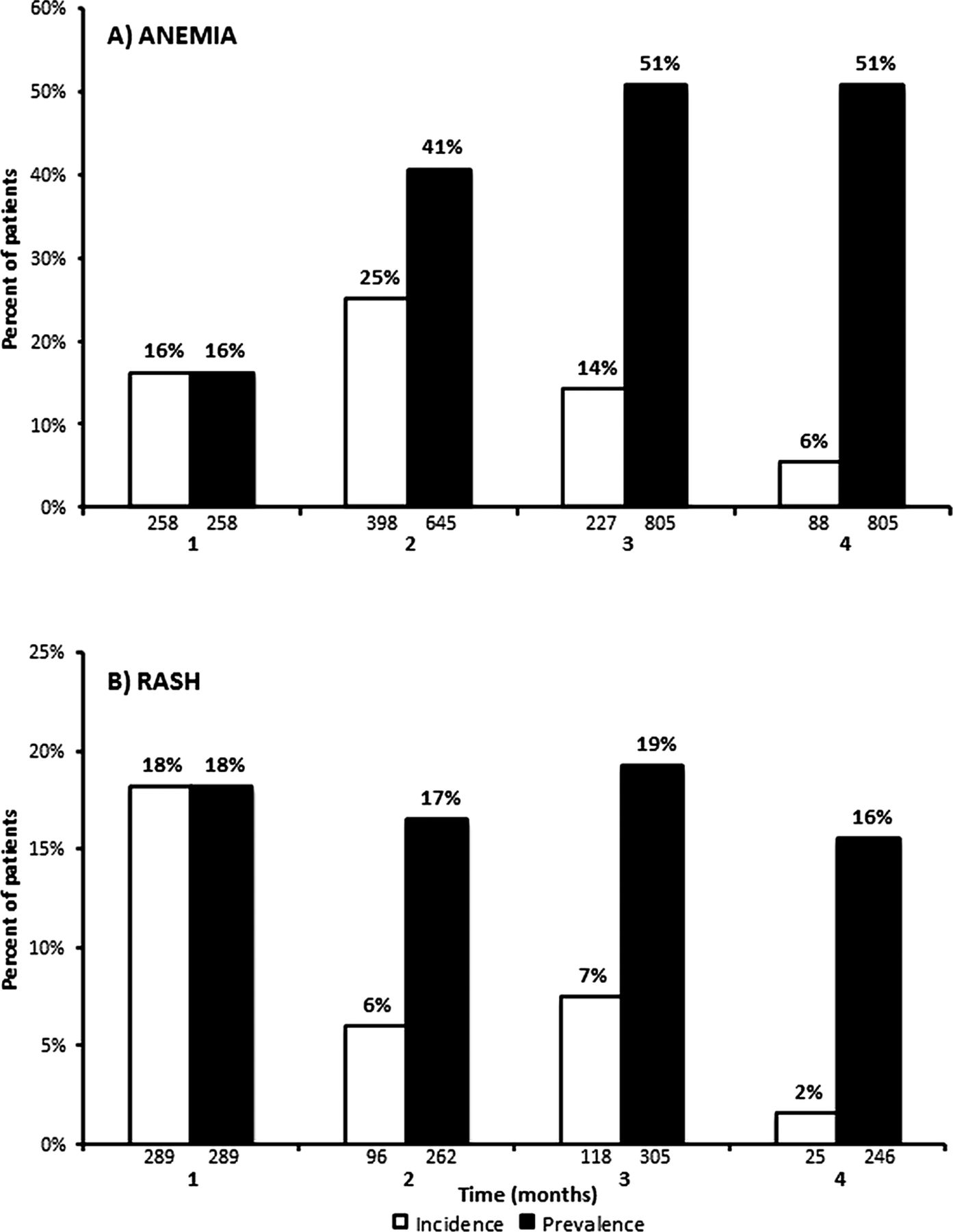
Figure 2 shows the incidence and prevalence rate of any grade TVR-related anaemia over the 16-week period of study. In all, 68% of cases of anaemia occurred within the first 8 weeks of treatment, and 83% of patients who developed anaemia throughout treatment were still considered anaemic at week 16.
Table 4 shows the different levels of anaemia recorded during the first 16 weeks of treatment, by baseline fibrosis stage. Overall, 45 patients (3%; 31 cirrhosis and 14 bridging fibrosis) discontinued TVR for anaemia, while 27 patients (2%) discontinued R for anaemia.

Figure 2
Incidence and prevalence of any grade telaprevir (TVR)-related anaemia (A) and rash (B). Shown are the incidence and prevalence rates of the Intent to Treat population by month from start of TVR treatment.
There were reductions in Hb below 10 g/dL in 48% of patients. In a multivariate analysis, the four strongest predictors of Hb <10 g/dL at any time on treatment were female sex (OR=1.69, 95% CI 1.27 to 2.27, p=0.0004), age>65 years (OR=2.31, 95% CI 1.46 to 3.65, p=0.0003), low baseline Hb (OR=1.08, 95% CI 1.06 to 1.09, p<0.0001) and higher weight-based dosing of R (OR=1.13, 95% CI 1.05 to 1.21, p=0.0005). Baseline Fibroscan test results were not a significant predictor of anaemia. There was no association between anaemia and the type of PEG-IFN.
By week 16, 201 patients (13%) had developed a grade 2–4 cutaneous rash that was considered drug-related; these figures were similar between patients with bridging fibrosis and those with cirrhosis (90; 12% vs 111; 13%). Of these, 64 (4%) were grade 3/4, and 28 cases (2%) were considered serious AEs including one patient who developed Stevens–Johnson syndrome, which resolved after stopping treatment. Overall, 72 patients (5%) discontinued drug treatment for rash of whom 36 had bridging fibrosis and 36 had cirrhosis at baseline (
table 2).
Figure 2 shows the incidence and prevalence of any grade TVR-related rash which, like anaemia, occurred more frequently during the first 8 weeks of therapy (73% of cases). Of patients who developed rash throughout treatment, 47% of cases were not resolved by week 16.
While the incidence of grade 2–4 infections considered treatment-related was low (n=14, 1%), 26 patients (2%) developed an infection as a serious AE, including seven cases of pneumonia, two cases of erysipelas, and one case each of sepsis and septic shock. Only four patients (<1%) discontinued TVR for infection.
DiscussionThe week 16 interim analysis of this EAP provided meaningful insights on the safety profile and on treatment efficacy of triple therapy with TVR in patients with histologically advanced hepatitis C, a category of patients who are more likely to suffer treatment-related AEs and to respond less satisfactorily to IFN-based regimens. Indeed, 12% of the 1587 patients with either bridging fibrosis or cirrhosis had to prematurely discontinue treatment owing to the onset of AEs, whereas grade 2–4 treatment-related anaemia or a cutaneous rash occurred in 44% and 13% of the patients, respectively, in the face of 82% of the overall cohort achieving on treatment clearance of serum HCV-RNA. Cirrhotic patients experienced slightly more grade 2–4 AEs than did patients with bridging fibrosis.
Owing to the multifactorial origin of anaemia in patients with advanced liver fibrosis including age, myelosuppression and impaired renal function, higher rates of grade 3 or 4 anaemia were observed in EAP patients than those observed in phase II/III studies (31% in the EAP compared with 4%–18% previously reported).
11 ,
12 ,
15 ,
20 ,
21 These discrepancies can be accounted for by the fewer number of patients enrolled in clinical development studies who had either bridging fibrosis or cirrhosis (ie, an advanced liver disease entailing a significant risk of developing myelosuppression-related AEs). Additionally, our choice of defining anaemia as either Hb below given threshold levels or any decrease of Hb following triple therapy could have increased the estimate of anaemia in some patients. In all, 40% of the overall cohort had anaemia managed with R dose reduction and blood transfusion was used in 10%. In contrast to the registration trial protocols where the use of EPO to correct anaemia was not permitted,
11 ,
12 332 patients (21%) received EPO. In addition, a significant proportion of patients had their anaemia treated through a combination of R dose reduction/interruption and either EPO or blood transfusion. It is possible that R dose reduction to manage anaemia was avoided because of concerns that treatment effectiveness may be compromised; however, it is now appreciated from both a retrospective and prospective study with TVR and BOC, respectively, that R dose reduction may not affect sustained virological response (SVR) rates.
22 ,
23 Overall, the strategies of anaemia management in this study resulted in only 3% of patients discontinuing TVR because of anaemia. This compares with 2% of patients in the REALIZE study who discontinued TVR for anaemia.
12Similar rates of anaemia were reported in patients with compensated cirrhosis who were enrolled in the field practice study CUPIC in France, where 29% had grade 3 anaemia (defined as Hb <9 g/dL);
15 this was despite a higher number of CUPIC patients (33%) with severe liver impairment exceeding the enrolment criteria adopted in REALIZE than in the EAP (9%).
12 The clinical burden of anaemia in CUPIC was higher than in the EAP, with EPO use in more than half of the patients (57%) or the need for blood transfusion in 15% possibly reflecting slightly more advanced liver disease in CUPIC patients.
Not unexpectedly, anaemia defined as a Hb drop below 10 g/dL following PR+TVR therapy more often occurred in >65-year-old patients, women and patients with low pretreatment Hb values or those with higher weight-based dosing of R. While age is a well-recognised risk factor for anaemia in patients with advanced liver disease, likely reflecting an increased susceptibility to treatment-related bone marrow and renal toxicity, female gender and low pretreatment Hb values have long been recognised as risk factors for anaemia also in patients exposed to dual therapy with PR.
24 ,
25One reassuring finding of the TVR EAP was the low rate of cutaneous rash (13% grade 2–4 treatment-related), and in particular of grade 3–4 rash affecting 4% of the population only. This may reflect the improved standard of care based on interventions and counselling to prevent cutaneous toxicity of TVR. This is not unprecedented since the rates of any grade rash were already reduced in phase III registration trials compared with the phase II trials where rash was a leading AE causing a shortened period of TVR administration.
15 ,
26 Similarly, rather low rates (4.8%) of grade 3/4 rash were also observed in the CUPIC study.
15In the EAP, a number of patients developed AEs other than anaemia or rash, including infections, thrombocytopenia, pruritus, weakness, nausea and anorectal discomfort. Similar to the cutaneous rash, anorectal discomfort was reported for a lower proportion of patients than in registration trials,
11 ,
12 ,
20 ,
21 ,
26 again possibly reflecting improved standard of care based on the use of prophylactic and therapeutic remedies. Ultimately, only 12% of patients experienced SAEs including anaemia (5%), rash (2%), infection (2%) and pyrexia (1%). Thus, enrolment of patients fulfilling the selection criteria to the registration trials and well compensated liver status resulted in a satisfactory safety record of TVR-based regimens in patients with advanced liver fibrosis. This explains also the low mortality rates (n=7, 0.4%) in the EAP cohort up to week 16. Deaths were the consequence of multi-organ failure caused by infection in two patients, pneumonia in two patients, and septic shock, ischaemic colitis, and haemorrhage in one patient each. Notably, one of these patients suffered from diabetes, a disease known to increase the risk of infection, and six of the seven patients had cirrhosis. PR-related deaths were reported also in patients with cirrhosis due to HCV (2%) who were enrolled in a trial aiming to evaluate the safety and efficacy of elthrombopag,
27 a synthetic compound able to increase the level of circulating platelets. Slightly higher rates (2.8%) of TVR treatment-related deaths were reported in patients with cirrhosis who were enrolled in the CUPIC study
15 where mortality was almost invariably associated with liver failure and predicted by signs of impaired liver function including a baseline platelet count less than 100 000/mm3 and serum albumin lower than 3.5 g/dL. In this study, TVR-based regimens were also associated with a significant rate of infection (6.5%). With respect to the TVR EAP, the greater burden of AEs and serious complications (29 among 429 patients) observed in the CUPIC study during the first 16 weeks of triple therapy underscore the enrolment of a significant number of patients with deteriorated liver function. In the French study, 20% of patients had <100 000 platelets and 12% <3.5 g albumin compared with TVR EAP where these figures were 8% and 1%, respectively. Poor safety signals in patients with more profound liver derangement were reported in two studies in Germany
16 and Austria
28 where the prevalence of patients with severe liver impairment was intermediate between CUPIC and TVR EAP.
The finding that 85% of previously untreated patients had undetectable HCV RNA at week 12 with minor differences between patients with bridging fibrosis and those with cirrhosis (87% vs 83%) is similar to the antiviral efficacy of TVR regimens seen in registration trials. Based on previous phase III trial results, one may therefore predict that a significant proportion of previously untreated patients with advanced fibrosis will ultimately achieve an SVR upon completion of the treatment schedule.
11 ,
21 Still, the SVR results need to be confirmed in long-term follow-up. In most countries, treatment naive patients with advanced fibrosis have been prioritised to receive triple therapy with TVR. The EAP study also provides encouraging though preliminary data of the efficacy of TVR triple therapy in treatment experienced patients with bridging fibrosis and cirrhosis. These findings were particularly rewarding for those individuals with a previous relapse or partial response to PR, reaching very high HCV RNA undetectability rates of 88% and 80% at week 12. A preliminary report of the CUPIC study is in line with our observation and predictions of SVR in experienced patients with advanced fibrosis/cirrhosis, with 46% of 107 patients enrolled in the study achieving an SVR at week 12 post-treatment.
22 One important limit of the CUPIC study, however, is the under-representation of null responders, who are most difficult to cure, as a consequence of their poor IFN sensitivity, leading to a high risk of virological breakthroughs and post-treatment relapse of hepatitis. By enrolling 436 patients who had had a prior null response (180 bridging fibrotic and 256 cirrhotic patients), the EAP is the largest study of prior null responders.
In conclusion, the 16-week interim analysis of the TVR EAP in 1587 patients provided encouraging insights on the safety, tolerability and preliminary efficacy of TVR triple therapy in difficult to cure categories of hepatitis C patients with advanced fibrosis.
AcknowledgmentsWe would like thanks the patients, investigators and staff who worked on this study.
FootnotesContributors Analysis and interpretation of data, drafting the article and revising it critically for important intellectual content: MC and HW. Conception and design: AV, WI, RD, AH, JML and IL-D. Statistical analysis: AH. Final approval of the version to be published: MC, IF, DA, PAF, SIS, PU, CM, AS-C, AV, WI, RD, AH, JML, IL-D and HW.
Funding This study was funded by Janssen Pharmaceutics.
Competing interests MC—Grant and research support: Merck, Roche, BMS, Gilead Science. Advisory committees: Merck, Roche, Novartis, Bayer, BMS, Gilead Science, Tibotec, Vertex, Janssen Cilag, Achillion, Lundbeck, Abbott, Boehringer Ingelheim. Speaking and teaching: Tibotec, Roche, Novartis, Bayer, BMS, Gilead Science, Vertex. IF—Janssen: Speakeŕs bureau; Gilead, MSD, Roche: Speakeŕs bureau. DA—Speaker and had grants for lectures and clinical trials from Merk, Jannsen, Roche, Novartis, BMS. PAF—Speaker and Clinical Investigator to Roche, Abbvie, Janssen, BMS. SIS—Received honoraria from Janssen for speaking and teaching and participating in Advisory Boards. PU—No conflicts of interest regarding the topic of manuscript. CM—Speaker or adviser from MSD, Janssen, Bristol-Myers Squibb and Gilead Sciences pharmaceutical companies. He is investigator for Novartis, Roche, MSD, Gilead Sciences, Bristol-Myers Squibb, Boehringer, Glaxo-Smith-Kline, Abbott, Astellas and Janssen pharmaceuticals. He received research Grant from MSD, Janssen, Astellas, Novartis and Roche pharmaceutical companies. AS-C—PI for the Telaprevir EAP-CT for subjects with Chronic Hepatitis C VX-950HEP3002; speaker for Janssen on different occasions. HW—Honoraria for consulting and/or lectures: Abbvie, Abbott, Achillion, BMS, Gilead, Janssen, MSD, Novartis, Roche, Roche Diagnostics, Siemens, Transgene. Grant support: Roche, BMS, MSD. AV, WI, RD, AH, JML and IL-D—Are Janssen employees.
Patient consent Obtained.
Ethics approval Local ethic committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing The principal investigator had full access to the results of the trial.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See:
http://creativecommons.org/licenses/by-nc/3.0/
ReferencesGhany MG,
Strader DB,
Thomas DL,
et al. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009;49:1335–74.
[CrossRef][Medline][Web of Science] ↵ European Association for the Study of the Liver (EASL). EASL clinical practice guidelines: management of hepatitis C virus infection. J Hepatol 2011;55:245–64.
[CrossRef][Medline][Web of Science] ↵Ghany MG,
Nelson DR,
Strader DB,
et al. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011;54:1433–44.
[CrossRef][Medline][Web of Science] ↵Dusheiko G,
Wedemeyer H. New Protease inhibitors and direct-acting antivirals for hepatitis C: interferon's long goodbye. Gut 2012;61:1647–52.
[FREE Full text] ↵Sarrazin C,
Hezode C,
Zeuzem S,
et al. Antiviral strategies in hepatitis C virus infection. J Hepatol 2012;56(Suppl 1):S88–100.
[Medline] ↵ Pearlman BL,
Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011;52:889–900.
[Abstract/FREE Full text] ↵ Aghemo A,
Lampertico P,
Colombo M. Assessing long-term treatment efficacy in chronic hepatitis B and C: between evidence and common sense. J Hepatol 2012;57:1326–35.
[CrossRef][Medline] ↵Van der Meer AJ,
Veldt BJ,
Feld JJ,
et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012;308:2584–93.
[CrossRef][Medline][Web of Science] ↵ Bourlière M,
Khaloun A,
Wartelle-Bladou C,
et al. Future treatment of patients with HCV cirrhosis. Liver Int 2012;32(Suppl 1):113–19.
[CrossRef][Medline] ↵Poordad FF,
McCone J,
Bacon BR,
et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011;364:1195–206.
[CrossRef][Medline] ↵Jacobson IM,
McHutchison JG,
Dusheiko G,
et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011;364:2405–16.
[CrossRef][Medline][Web of Science] ↵Zeuzem S,
Andreone P,
Pol S,
et al. Telaprevir for retreatment of HCV infection. N Engl J Med 2011;364:2417–28.
[CrossRef][Medline][Web of Science] ↵Bacon BR,
Gordon SC,
Lawitz E,
et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011;364:1207–17.
[CrossRef][Medline] ↵Gao X,
Stephens JM,
Carter JA,
et al. Impact of adverse events on costs and quality of life in protease inhibitor-based combination therapy for hepatitis C. Expert Rev Pharmacoecon Outcomes Res 2012;12:335–43.
[CrossRef][Medline] ↵Hézode C,
Fontaine H,
Dorival C,
et al. Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multicentre cohort of the French Early Access Programme (ANRS CO20-CUPIC)—NCT 01514890. J Hepatol 2013;59:434–41.
Search Google Scholar ↵Maasoumy B,
Port K,
Markova AA,
et al. Eligibility and safety of triple therapy for hepatitis C: lessons learned from the first experience in a real world setting. PLoS ONE 2013;8:e55285.
[CrossRef][Medline] ↵ Telaprevir (INCIVEK) Package Vertex Pharmaceuticals. http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/201917lbl.pdf (accessed October 2013).
↵ Castéra L,
Verginol J,
Foucher J,
et al. Prospective comparison of transient elastography, Fibrotest, APRI, and liver biopsy for the assessment of fibrosis in chronic hepatitis C. Gastroenterology 2005;128:343–50.
[CrossRef][Medline][Web of Science] ↵ DAIDS (Division of AIDS). Table for grading the severity of adult and pediatric adverse events. Bethesda, MD, USA: DAIDS, 2004.
↵Hezode C,
Forestier N,
Dusheiko G,
et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med 2009;360:1839–50.
[CrossRef][Medline] ↵Sherman KE,
Flamm SL,
Afdhal NH,
et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011;365:1014–24.
[CrossRef][Medline][Web of Science] ↵Sulkowski MS,
Roberts S,
Afdhal N,
et al. Ribavirin dose modification in treatment-naïve and previously treated patients who received telaprevir combination treatment: no impact on sustained virologic response in phase 3 studies. J Hepatol 2012;56(Suppl 2):S459–60.
Search Google Scholar ↵ Poordad FF,
Lawitz EF,
Reddy KR,
et al. A randomized trial comparing ribavirin dose reduction versus erythropoietin for anemia management in previously untreated patients with chronic hepatitis C receiving boceprevir plus peginterferon/ribavrin. J Hepatol 2012;56(Suppl 2):S559.
Search Google Scholar ↵Sulkowski MS,
Shiffman ML,
Afdhal NH,
et al. Hepatitis C virus treatment-related anemia is associated with higher sustained virologic response rate. Gastroenterology 2010;139:1602–11.
[CrossRef][Medline] ↵Ogawa E,
Furosyo N,
Nakamuta M,
et al. Clinical milestones for the prediction of severe anaemia by chronic hepatitis C patients receiving telaprevir-based triple therapy. J Hepatol 2013;59:667–74.
Search Google Scholar ↵McHutchison JG,
Everson GT,
Gordon SC,
et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009;360:1827–3.
[CrossRef][Medline] ↵Dusheiko G,
Afdhal N,
Giannini EG,
et al. Results of enable 2, a phase 3, multicenter study of eltrombopag and peginterferon alfa-2b treatment in patients with hepatitis C and thrombocytopenia. J Hepatol 2012;56:S21–44.
Search Google Scholar ↵Rutter K,
Ferlitsch A,
Maieron A,
et al. Safety of triple therapy with telaprevir or boceprevir in patients with advanced liver disease—predictive factors for sepsis. European Association for the Study of the Liver (EASL) Conference; April 2013. [abstract].
Search Google Scholar
 Dec 9 - Fair Pricing Coalition Condemns Gilead Sciences on the High Price of New Hepatitis C Drug Sovaldi™, and Urges Rapid and Wide Dissemination of Support Program Details for Uninsured and Underinsured People Living with Hepatitis C
Dec 9 - Fair Pricing Coalition Condemns Gilead Sciences on the High Price of New Hepatitis C Drug Sovaldi™, and Urges Rapid and Wide Dissemination of Support Program Details for Uninsured and Underinsured People Living with Hepatitis C