HCV Weekly Rewind
From Journal of Viral Hepatitis @ Medscape
Treating Hepatitis C Current Standard of Care and Emerging Direct-acting Antiviral Agents
F. Poordad; D. Dieterich
Posted: 06/29/2012; J Viral Hepat. 2012;19(7):449-464. © 2012 Blackwell Publishing
Abstract and Introduction
Abstract
During the late 1990s and early 2000s, major advances were made in the treatment of patients with chronic hepatitis C virus (HCV) infection. Interferon, combination interferon plus ribavirin (RBV) and pegylated interferon plus RBV increased sustained virologic response (SVR) rates from ~5% to ~40–80%, depending on the genotype of HCV infection. Advances in molecular biology have allowed investigators to begin to understand the mechanisms of HCV infection and replication. Advances in understanding of viral kinetics have provided tools to identify patients who are most likely to attain SVR. With the advances in the science of HCV infection, the first part of the 21st century has seen the development and early introduction of a number of direct-acting antiviral (DAA) drugs. These novel medications interfere with critical steps in HCV replication and have the potential to significantly increase SVR rates. This article will review the key elements of HCV replication and evaluate the various classes of new and investigational DAA that have the potential to create a revolution in the management of patients with chronic hepatitis C.
Introduction
Globally, at least 170 million people are chronically infected with the hepatitis C virus (HCV), with 9 million infected in the United States and Western Europe.[1,2] The prevalence of the infection varies geographically, from 0.1% to 5%.[3] The highest rates are reported in Africa and the Eastern Mediterranean region.[4] Since 1982, the incidence of acute HCV infection in the United States has fallen from 7.4/100 000 to 0.7/100 000, except in human immunodeficiency virus (HIV) seropositive men who have sex with men (MSM), for whom it is increasing dramatically.[5] This decline is predominantly attributed to HCV screening in transfusion centres, and concerns about HIV infection that have led to institution of universal precautions in healthcare settings and changes in behaviour of injection drug users.[6] Despite the fall in incidence, the number of patients with complications of chronic hepatitis C is increasing.[7] Currently, 40–50% of all liver transplants in the United States are in complications of HCV infection.[8] The reason for this is that chronic HCV infection is usually silent and hepatic fibrosis, cirrhosis, end-stage liver disease and hepatocellular carcinoma develop only after a decade or more of chronic liver injury and hepatic remodelling. The burden of chronic hepatitis C is expected to increase well in the future and continue to be an important cause of premature mortality.[7,9]
Evolution of the Standard of Care
At the present time, chronic hepatitis C is the only chronic viral infection that can be cured with antiviral therapy. Unlike HIV or hepatitis B (HBV), HCV nucleic acids are not integrated into the host genome, nor is there a viral latency phase. Although prevention of infection should be a primary goal, no HCV vaccine is available. Therefore, the aim of the current anti-HCV approaches is to cure the infection to prevent HCV-related complications and potential spread to others. The standard of care for the past decade and the comparator with newer therapies is a combination of pegylated interferon (PegIFN) plus ribavirin (RBV). Standard interferon was approved for HCV treatment in 1990; it represented a significant advance since up to then there was no treatment available, but the sustained virologic response (SVR) was <10% in patients with HCV genotype 1 (Table 1).[10]
Table 1. Therapeutic response parameters
| Parameter | Comments |
|---|---|
| Rapid virologic response (RVR) | RVR: Negative hepatitis C virus (HCV) RNA after 4 weeks of treatment |
| Extended RVR | eRVR: RVR that remains negative at 12 weeks |
| Early virologic response (EVR) | EVR: Reduction in HCV RNA ≥2 log10 (partial EVR) or negative (complete EVR) at 12 weeks |
| Delayed virologic response (DVR) | DVR: Patients who fail to achieve EVR but are HCV negative at 24 weeks |
| End-of-treatment (EOT) response | EOT: HCV RNA negative at the EOT |
| Relapse | HCV RNA negative at EOT with viraemia reappearing when treatment is stopped |
| Sustained virologic response (SVR) | SVR: Negative HCV RNA by PCR 6 months after EOT |
In 1998, combination therapy of interferon plus RBV increased SVR rates by another 14–22%. The pharmacokinetics of standard interferon, which required thrice weekly dosing, was subsequently improved by pegylation of the molecule, allowing once weekly dosing, thereby increasing SVR rates to ~50% in patients with HCV genotype 1.[11,12] Although two forms of the recombinant interferon have been marketed (peginterferon alpha 2a and 2b), the rates of response and tolerability of the two drugs are equivalent: that is, neither formulation of PegIFN nor dosing (low vs conventional for peginterferon alpha-2b) significantly alters SVR rates or tolerability.[13] Weight-based RBV remains indispensable to the treatment of HCV and will be for the foreseeable future. Response does vary by genotype. Because genotypes 1 and 4 are less responsive than are genotypes 2 and 3, the SVR rates by genotype with pegIFN/RBV combination therapy (PR) ranged from 40% to 50% for the former and ~80% for the latter.[11,12] Thus, although PR has been the current standard of care (SOC) and the comparator often used in clinical trials of developmental agents, the low SVR in genotype 1 and the very poor response rates in subpopulations such as black patients and cirrhotics have driven the development of novel antiviral therapies.
HCV Infection and Replication
The infection and replication processes utilized by the HCV provide potential therapeutic targets (Fig. 1). Once the virus enters the bloodstream, HCV associates with low-density and very low-density lipoproteins (LDL, VLDL), and the virus's E1 and E2 structural proteins subsequently bind to receptors on hepatocytes.[14] Proposed binding sites include the LDL receptor (LDLR), hepatocyte glycosaminoglycans, CD81, scavenger receptor-BI (SR-BI) and claudin-1.[15–18] The virus's highly glycosylated envelope fuses with the hepatocyte's cell membrane and subsequently undergoes clathrin-mediated endocytosis.[19] Acidification of the vesicle frees the positive-strand genomic RNA from the nucleocapsid and releases the virus into the cytoplasm.[20] Along with host mRNA molecules, viral RNA migrates to the rough endoplasmic reticulum (rER). Binding of the viral RNA's 5′ untranslated region's internal ribosome entry site (IRES) with a 40S ribosomal subunit forms a stable pre-initiation complex that begins translation of the viral genome to form the HCV polyprotein.[21]
Figure 1.
Hepatitis C virus infection and replication – potential therapeutic targets. Reprinted with permission from Macmillan Publishers Ltd.25

Following translation, the ~3,000-amino-acid HCV polyprotein is co- and postprocessed by host peptidases and viral proteases into at least 11 viral proteins (Fig. 2).[22,23] Viral proteins participate in the formation of a complex of small vesicles derived from the ER and embedded in a matrix known as the membranous web.[24] Replication is largely mediated by nonstructural protein 5B (NS5B), an RNA-dependent RNA polymerase (RdRp); however, NS5A also participates in the operation. In a 2-step process, positive-strand genomic RNA serves as a template for synthesis of a negative-strand intermediate. Once synthesized, the negative strands of RNA then also act as a template to produce numerous copies of positive-strand genomic RNA that can either be translated into new HCV polyproteins or packaged and released as new viral particles. Within the ER, core protein interacts with the positive-stranded HCV RNA, while the envelope proteins are glycosylated and the components are assembled into new virions. New viruses then bud into the ER or an ER-derived compartment and then exit the cell through a secretory pathway.[25]
Figure 2.
Genomic organization of hepatitis C virus polyprotein. Adapted from Uprichard.23

.
Hepatitis C virus infection is a dynamic process.[25] The virus half-life is only a few hours, and an infected patient may make and clear 1012 virions/day.[26] As a result of the high replicative activity, existence of hypervariable regions within the HCV genome, and the fact that RdRp lacks proofreading ability, there is a tremendous diversity in an individual (quasispecies) or an infected population.[25] Over centuries, HCV strains have evolved, so that the nucleotide sequences of various strains differ by as much as 30–35%. This allows them to be subtyped into one of six genotypes and several subtypes. HCV genotypes are distributed by geographic region and influence the outcomes of current combination PR therapy. These viral kinetic properties are similar to those of HIV. Many of the copying errors will produce replication-deficient or nonfunctional HCV genomes. However, heterogeneity within the individual also creates HCV subpopulations with a survival advantage. Because of selection pressure exerted by the host's immune system and/or treatment, viruses with 'hearty' genomes have a survival advantage and can become the dominant strain.[27] These considerations provide a strong rationale for the development and implementation of antiviral combination therapies.Although not the focus of this review, HCV resistance is a major concern with newer and investigational agents. Variants that exist naturally are present at the initiation of treatment and emerge during therapy in the setting of poor interferon response.
Direct-acting Antivirals in Development for HCV
Until recently, the lack of a suitable in vitro model of HCV replication has limited development of new drugs to treat chronic hepatitis C. Although the virus can infect and replicate in chimpanzees, use of non-human primates for drug development is limited by a number of factors including cost, housing and need for regular veterinary care. In 1999, Lohman et al.[28] constructed an HCV replication system from genotype 1b RNA and the human hepatoma cell line Huh-7. Subsequently, a cloned HCV genome was developed that recapitulates the virus's full life cycle in cell culture and produces acute hepatitis C in chimpanzees.[29] This has facilitated screening of large numbers of potential anti-HCV drugs and identification of a number of potential direct-acting antiviral (DAA) candidates. These agents target specific genomic pathways that can interfere with HCV infection and replication.
Genomic Targets
Potential genomic targets of DAA include both structural and nonstructural proteins (Fig. 2).[23,30] Structural proteins that could serve as targets include the nucleocapsid core protein (C) and envelope glycoproteins E1 and E2. Nonstructural proteins targeted include the NS2/NS3 region, the NS3 serine protease RNA helicase, the NS4A peptide cofactor of NS3, the NS5A protein and the NS5B RNA-dependent RNA polymerase.
HCV Envelope Proteins
E1 and E2 are proteins embedded in the virus's lipid envelope.[25] They are essential to the HCV life cycle because they are required for the virus to gain entry into the host hepatocyte's cytoplasm. Envelope proteins are located between the C and P7 structural proteins on the amino-terminus end of the polyprotein. Inhibition of the envelope proteins could prevent the virus from gaining access to the cytoplasm and decrease the ability of newly synthesized HCV to infect HCV-naïve hepatocytes. However, hypervariable regions of E2 suggest that immune pressure might select escape variants if envelope proteins are targeted with either a vaccine or a DAA.[24]
P7 Protein
This viral protein is located between the E2 and NS2 regions of the HCV polyprotein, which is the interface between the HCV structural and nonstructural coding regions.[31] Experimentally, P7 can be identified and associated with the ER, mitochondria and cell membrane. In artificial lipid membranes, P7 has ion channel activity. In a non-human primate model, RNA transcripts with P7 mutations were not infective.[32]
NS2/NS3 Proteins
NS2 is the first nonstructural protein on the amino-terminal end of the polyprotein.[33] Before it splits from the polyprotein, NS2 forms a dimeric proteinase complex with NS3 that autocatalytically separates the two.[31] Although it is not essential for the formation of the replication complex, NS2 protein participates in virus assembly and release. Inhibition of NS2 blocks processing of the HCV polyprotein. NS3 participates in the NS3A/NS4B protease activity. The amino-terminal third of NS3 has serine protease activity, whereas the remaining portion of the protein shows RNA helicase properties. When expressed alone, NS3 is diffusely distributed throughout the hepatocyte's cytoplasm.[31]
NS3/N4A Protease
NS3 forms a complex with NS4A, a small protein that functions as a serine protease cofactor. When coexpressed, the two nonstructural proteins are localized to ER and ER-like membranes.[31] Together, they are responsible for cleaving the nonstructural proteins that are located downstream of NS3 towards the polyprotein's carboxy-terminus. NS4A helps protect the NS3/NS4A from proteolytic degradation and also participates in blocking elements of the hepatocyte's innate immune response.
NS4B Protein
Another downstream nonstructural domain of the HCV polyprotein is NS4B. Although its function is not understood completely, the protein appears to participate in the formation of the RNA replication complex.[31] Domains of the NS4B protein can be identified on both the internal and external surfaces of the ER. Localized there, the protein may function in crosstalk between the lumen of the ER and the cytosol.
NS5A Protein
Once cleaved from the polyprotein, NS5A localizes to membranes.[31] There, it binds to the 3′ end of the virus's newly produced positive- and negative-stranded RNA and participates in genome replication. Mutations of NS5A have the potential to markedly enhance HCV replication. NS5A interacts with numerous cellular signalling pathways, including growth, apoptosis and interferon production.[33] The protein also interacts with NS5B to maintain HCV replication.
NS5B Polymerase
Like most HCV proteins, NS5B is associated with the ER or ER-derived membranes.[31] The protein is RdRp, a principal component of the HCV replication apparatus. Other HCV proteins such as NS3 and NS5A modulate its activity. The RNA affinity of the NS5B is upregulated by the host cell's cyclophilin B.
Class Properties of Direct-acting Antiviral Drugs
Both the therapeutic targets and mechanisms of action are used to classify the DAA. As such, the various classes share some similarities (Fig. 3). NS3/4A inhibitors block the enzymatic activity required to cleave HCV into individual structural and nonstructural proteins and are known as protease inhibitors (PIs).[34] On the basis of the time they were approved, potency, activity against resistant strains, bioavailability, safety and tolerability, PIs have been grouped into first- and second-generation agents, and to first- and second-wave within each generation. For example, agents with a similar resistance profile are considered to be of the same generation and could be grouped into separate waves depending on dosing, safety and tolerability characteristics. Although the exact mechanism of action is unknown, the NS5A protein is essential for replication and assembly of HCV. Inhibitors of NS5A block viral production at an early stage of assembly, so that no viral RNA or nucleocapsid protein is released.[35] Agents that block NS5B activity inhibit the virus's RdRp and are referred to as polymerase inhibitors.[34] Both nucleoside and non-nucleoside RdRp inhibitors have been synthesized. Nucleoside inhibitors (NIs) bind to RdRp's active site, whereas the non-nucleoside inhibitors (NNIs) bind to the enzyme outside the active site, inducing conformational changes that downregulate RdRp's activity. As a result of mechanistic and potency differences, the NIs tend to have broad potency against multiple HCV genotypes and are less likely to select for resistant strains than are the NNIs.[36] Cyclophilin is a host protein that interacts with NS5B and appears to promote the HCV protein's ability to bind to viral RNA.[37]
Figure 3.
Direct-acting antiviral agents

Licensed Direct Antivirals
First-generation NS3/4A Protease Inhibitors
Telaprevir Telaprevir (VX-950; trade name Incivek, Vertex Pharmaceuticals, Cambridge, MA, USA) and boceprevir (SCH503034; trade name Victrelis, Merck & Co., Inc., Whitehouse Station, NJ, USA) constitute the first wave of the first generation of PIs. Because several patients in phase I trials with telaprevir had viral breakthrough during telaprevir treatment, subsequent clinical trials have combined the PI with PR.[38–40] Telaprevir has been studied in three phase III trials: A New Direction in HCV Care (ADVANCE), Re-treatment of Patients with Telaprevir-Based Regimen to Optimize Outcomes (REALIZE) and ILLUMINATE.
The ADVANCE study evaluated telaprevir-based regimens in 1088 treatment-naïve patients infected with HCV genotype 1 who were randomized to either PR for 48 weeks, or telaprevir 750 mg Q8h for either 8 (T8/PR) or 12 (T12/PR) weeks plus a full 48 weeks of PR.[41] An eRVR was achieved by 57% and 58% of patients in the T8/PR and the T12/PR arms, respectively, whereas only 8% of those receiving the standard PR regimen achieved eRVR. Those who achieved an eRVR stopped treatment at 24 weeks; those who did not but had negative HCV RNA at week 24 continued PR for the full duration. Compared with PR, subjects who were randomized to the T8 or the T12 regimen achieved SVR of 69% and 75%, respectively, vs 44% with SOC. On-treatment virologic breakthrough occurred less often with the 12-week regimen (8%vs 13%).
REALIZE evaluated the first-generation PI in 663 patients with HCV genotype 1 infection who had previously failed PR.[42] Enrollees included nonresponders (n = 184), partial responders (n = 124) and relapsers (n = 354). Patients were randomized to one of three arms: T12/PR48, PR4 (lead-in) + T12/PR40 or PR48. The SVR rates were similar in both telaprevir arms, with 64% in the T12/PR48 arm and 66% in the arm with the 4-week PR lead-in. After analysis of the data by the United States Food and Drug Administration, the manufacturer was allowed to report an adjusted SVR rate of 79% with 12 weeks of telaprevir plus PR. SVR rates in the telaprevir (both arms combined) vs conventional therapy arms were 31%vs 5%, 57%vs 15% and 86%vs 24% in the nonresponders, partial responders and relapsers, respectively. The manufacturer recommends that the drug be discontinued in all patients with HCV RNA levels of ≥1000 IU/mL at treatment week 4 or 12 or confirmed detectable HCV RNA levels at treatment week 24.[43]
ILLUMINATE was a noninferiority trial assessing response-guided therapy (RGT) in 540 treatment-naïve patients.[44] Those who achieved an eRVR were subsequently randomized at week 20 to PR for a total duration of 24 or 48 weeks. Of all enrollees, 65% achieved an eRVR, using a lower limit of quantification of 25 IU/mL. The SVR rates with PR for 24 weeks were noninferior to 48 weeks (92%vs 88%). In the ITT analysis, the overall SVR rate was 72% and the relapse rate was 7.7%. Treatment was discontinued in 8% and 18% of patients because of virologic failure and AEs, respectively. The latter were primarily anaemia and fatigue.
Like telaprevir, boceprevir (trade name Victrelis, Merck & Co., Inc.) is a first-generation PI for treatment of people with HCV genotype 1 infections. The HCV-SPRINT trials were phase III studies of boceprevir. HCV-SPRINT-2 evaluated the PI in 1097 treatment-naïve patients.[45] All enrollees received PR during a 4-week lead-in period and were then randomized to one of three arms: PR + placebo for an additional 44 weeks (PR/PBO); PR + boceprevir for an additional 24 weeks if undetectable by week 8, or followed by another 20 weeks of PR (PR/B24) for those becoming virus undetectable after week 8 but before week 24; or PR + boceprevir for an additional 44 weeks (PR/B44). Treatment was discontinued if the patients were HCV positive at 24 weeks. The SVR rates for PR/PBO, PR/B24 and PR/B44 were 40%, 67% and 68%, respectively, for nonblack subjects. The increases in SVR rates for the two boceprevir-containing regimens were statistically significant when compared with group 1. For black patients, the SVR rates were 23%, 42% and 53%, respectively. Although the differences were statistically significant for the two boceprevir-containing regimens vs PR, the null hypothesis was accepted for the two PI arms. It should be noted that boceprevir has not been studied in treatment-experienced patients who did not achieve an EVR with PR. On the basis of the analysis of the clinical trials, in-treatment HCV RNA levels can be used to predict the optimal duration of therapy (RGT) with discontinuations of treatment as early as 28 weeks. The FDA and the manufacturer recommend that boceprevir treatment be discontinued if HCV RNA levels are ≥100 IU/mL at week 12 or detectable at week 24.[46]
The RESPOND-2 trial (Fig. 4) assessed previously treated patients who had relapsed or had a nonresponse, but excluded historic 'null' responders, who had less than a 2-log decline by week 12 when treated with PR.[47] A lead-in paradigm was used to compare 48 weeks of PR with two different boceprevir treatment regimens: a standard PR4 + PR/B44 vs and RGT PR4 + PR/B32 +/− PR12 depending on week 8 viral response. The SVR rates in the RGT arm vs the PR4 + PR/B44 arm were 59%vs 64%, both of which were superior to the 21% SVR of PR.
Figure 4.
Respond-2 study design. Reprinted with permission from the Massachusetts Medical Society.47
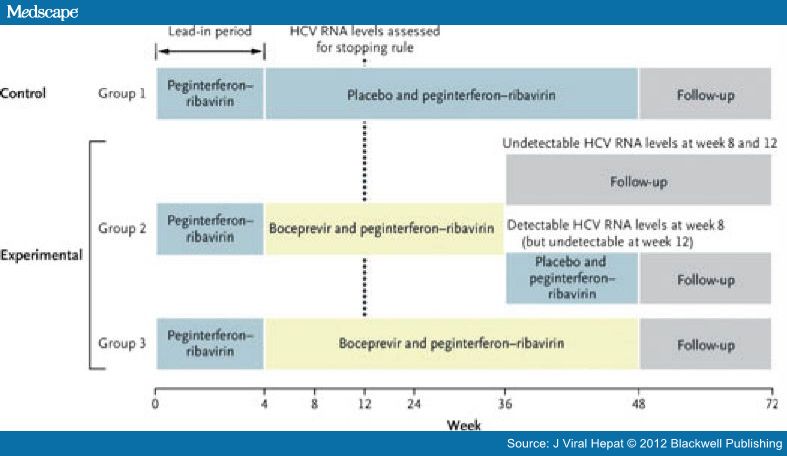
Although null responders were not permitted in the study, the lead-in identified that 26% of patients had a week 4 null response of <1 log decline, and these individuals had SVR rates of 33–34%, very similar to the response of 12-week null responders in the telaprevir studies. Hence, the labelled indication for boceprevir also includes null responders, who should receive PR4 + PR/B44.[46] This so-called classic null population was subsequently studied in the PROVIDE trial and achieved end-of-treatment response of 41%, with a projected SVR in the 30% range.
Agents Undergoing Clinical Trials
Second-wave, First-generation Protease Inhibitors
The second wave of PIs for HCV includes agents with improved potency and dosing, but with resistance profiles that are similar to telaprevir and boceprevir. It is proposed that the term 'second-generation' PI be used for agents that have an improved resistance profile, such as several that are currently in development.
TMC435
TMC435 (Tibotec, Medivir Pharmaceuticals, Janssen, Raritan, NJ, USA) is a potent, noncovalent HCV NS3/4A protease inhibitor. In a phase IIa, double-blind, placebo-controlled trial of a treatment-naïve and treatment-experienced genotype 1 population, patients were randomized to two groups with a total of seven arms. The first group received TMC435 (25, 75 or 200 mg QD) or placebo (PBO) as a loading dose for a week followed by another 3 weeks of the PI and 24–48 weeks of PR. The second group received 4 weeks of the PI (25, 75 or 200 mg QD) or PBO in combination with PR for either 24 or 48 weeks. Viral load reduction in subjects in the second group who received the PI at 25, 75 or 200 mg QD with concurrent PR were −4.74, −5.52 and −5.44 log10 units, respectively. Grade 3/4 AEs were reported in 10–11% of subjects who received the 200-mg daily dose. The most common AEs were fatigue, nausea, influenza-like symptoms and headache. At 12 weeks, all patients who received the PI (75 or 200 mg) for 4 weeks + concurrent PR had HCV RNA <10 IU/mL.
The PILLAR study (Protease Inhibitor trial assessing the optimaL dose and duration as once daiLy Anti-viral Regimen) is an ongoing, 5-arm, global phase IIb randomized, double-blind, placebo-controlled study in 386 treatment-naïve patients. TMC435 was administered in doses of 75 mg or 150 mg QD for either 12 or 24 weeks in combination with 24 weeks of PR. Patients receiving TMC435 were allowed to stop all treatment at week 24 with HCV RNA levels <25 IU/mL at week 4 and HCV RNA <25 IU/mL at weeks 12, 16 and 20. Patients who did not meet criteria for a virologic response continued with PR for the full 48 weeks. TMC435 demonstrated potent antiviral activity, at week 4 (RVR) and at week 12 (cEVR) with 92%. HCV RNA was undetectable (<25 IU/mL) for the majority of patients; 83% of them were able to stop all treatment at week 24. The viral breakthrough rate was 4.9% in those who received the PI. Interim 24-week results from the stage IIb ASPIRE trial of TMC435 100 mg QD or 150 mg QD in combination with PR have revealed similar results in treatment-experienced patients infected with HCV genotype 1 who had previously received at least 1 course of PR. At week 24, with the higher dose of TMC435, HCV RNA was undetectable (<25 IU/mL) in 94% of relapsers, 89% of partial responders and 87% of nonresponders.[48] However, it should be noted that approximately 90% of the patient population in ASPIRE was white. As previously described, decreased viral responses are observed in black populations as compared with nonblack populations, which suggests that the patient population in ASPIRE is easier to treat than is a more representative cross-section of the total HCV patient population. Phase III studies in various stages of execution will examine safety and efficacy of the PI in treatment-naïve patients, relapsers and nonresponders.
Asunaprevir
Asunaprevir (BMS-650032; Bristol-Myers Squibb, New York, NY, USA) is an HCV NS3 PI that is dosed orally at 600 mg twice daily and is currently undergoing phase two clinical studies. In an open-label, phase 2a study that was conducted with 21 nonresponder, genotype 1 patients, the combination of asunaprevir plus an NS5A inhibitor (daclatasvir) for 24 weeks was associated with an SVR at 12 and 24 weeks after treatment in two of nine patients with HCV genotype 1a and two of two patients with genotype 1b (i.e. 36% of the patients in this treatment arm).[49] Six patients (all genotype 1a) experienced viral breakthrough in this treatment arm. In the comparator treatment arm, patients received asunaprevir plus daclatasvir and PR. All 10 patients in the comparator arm achieved an SVR at 12 weeks after treatment, and nine achieved an SVR after 24 weeks of treatment. Although a significant proportion of the genotype 1a patients experienced breakthrough on the all-oral DAA dual combination treatment, this study demonstrated that an SVR can be achieved with an interferon-free regimen consisting of only two DAA.
BI 201335
BI 201335 (Boehringer Ingelheim Pharmaceuticals, Ridgefield, CT, USA) is an HCV NS3 PI given once daily currently in phase III. The phase IIb SILEN-C1 trial was conducted in patients with HCV genotype 1 naïve to HCV treatment to evaluate safety and efficacy of BI 201335 given once daily at a dose of 120 or 240 mg for 24 weeks in combination with PR for 24 vs 48 weeks.[50] A lead-in of PR for 3 days was also evaluated in two of the four arms (with 120 mg and 240 mg QD). In the two arms using BI 201335 at a dose of 240 mg (with and without a lead-in), patients achieving eRVR were re-randomized to either stop treatment at week 24 or continue with PR for a total of 48 weeks. Patients receiving 240 mg QD without a lead-in achieved the highest eRVR rate of 87% and were thus eligible for shortened treatment duration. As expected with this high eRVR rate, this arm also had the highest SVR rate of 83% vs 73% of patients receiving 240 mg with a lead-in and 56% of patients in the PR control group. Prolonging treatment to 48 weeks in those patients achieving eRVR did not result in higher SVR rates. Of those who completed 24 weeks, 93% achieved SVR vs 90% of those who completed 48 weeks. Viral breakthroughs occurred in 2.8–5.8% of patients receiving BI 201335 with the highest rate in those of the 120 mg daily with lead-in arm.
The phase IIb SILEN-C2 trial evaluated BI 201335 for 24 weeks in combination with PR for 24 vs 48 weeks, with or without a 3-day lead-in of PR in previous partial and null responders infected with HCV genotype 1.[51] The 240-mg QD dose (with and without a lead-in) was compared to 240 mg twice daily with a lead-in. Patients of the 240-mg QD group with lead-in achieving eRVR were re-randomized to stopping therapy or continuing 48 weeks with PR. Similar to the SILEN-C1 trial, the lead-in did not appear to improve efficacy. The 240-mg QD dosing without a lead-in led to the highest SVR rates. Overall, eRVR was achieved by 45% of patients, and SVR was achieved by 27–41% of patients. The lowest SVR rate was observed in the 240 mg QD with lead-in arm, the one group that used RGT for those achieving eRVR. In comparison with the good results observed with 24 weeks of treatment in the naïve patients achieving eRVR in the SILEN-C1 trial, prior partial and null responders achieving eRVR in SILEN-C2 achieved lower SVR rates when stopped at week 24. Only 40% of patients achieved SVR when stopped at week 24 compared to 72% of those who completed 48 weeks of treatment. The additional 24 weeks of PR greatly impacted the relapse rate. Sixty per cent (60%) of those who stopped at week 24 relapsed compared to 21% of those who completed 48 weeks of treatment. Viral breakthroughs occurred predominantly on BI 201335 compared to PR (17–28%vs 5–7%). Several adverse events were reported in a higher proportion of patients receiving BI 201335 compared to those on PBO and were dose dependent. Jaundice, skin manifestations, including rash, photosensitivity reactions, pruritus and dry skin, and gastrointestinal side effects, mostly nausea, vomiting and diarrhoea, were reported in the BI 201335 arms in a proportion exceeding 10% of the placebo/PR group. Jaundice was secondary to predominantly indirect or unconjugated hyperbilirubinaemia. This was dose dependent, rapidly reversible in all cases at cessation of BI 201335 and not associated with liver injury. The mechanism of action is inhibition of hepatic uptake of uridine diphosphate glucuronosyl transferase 1 family polypeptide A1 (UGT1A1).[52]
ACH-1625
ACH-1625 (Achillion Pharmaceuticals, New Haven, CT, USA) is a potent inhibitor of HCV NS3 protease that is a first-generation, second-wave PI. It is rapidly and selectively distributed to the liver.[53] In phase Ib clinical studies, HCV genotype 1-infected patients receiving doses ranging from 200 to 600 mg BID, and 400 to 600 mg QD for up to 5 days showed mean maximal reductions in viral load ranging from 3.1 1og10 to 4.25 1og10 (Fig. 5).[54] Reductions in viral load were substantial at 48 h. Furthermore, all patients had viral loads that remained suppressed for at least 7 days after dosing was completed, maintaining a mean reduction of more than 1.0 1og10 from baseline through day 12, the last day of viral load measurement in the study. In the 400- and 600-mg fasting and the 600-mg-fed groups, the maximum decreases in HCV RNA were 3.67, 3.40 and 3.81 log10, respectively. With QD dosing, modelling showed that the log drop does not fall below 3 and that there were no major differences between the QD and previously described BID dosing regimens. Despite a high proportion of resistant variants identified in participants and drug plasma level significantly below the EC50 against those strains, viral suppression continued through the follow-up at day 12.[55] This observation may be the result of the drug's hepatoselectivity.
Figure 5.
Mean antiviral response for 5-days of ACH-1625 and 7 days of follow-up.

Using data from the phase I trial, a single-population viral dynamics model was used to estimate the viral kinetics parameters of combination of ACH-1625 plus PegIFN.[56] When combined with PegIFN, ACH-1625 at 200 mg BID or 600 mg QD was predicted to increase Rapid virologic response (RVR) >2-fold as compared with PegIFN monotherapy. Viral kinetics parameter estimates obtained from modelling of ACH-1625 monotherapy were robust, and the dynamic modelling results suggest that this PI has the potential to enhance achievement of RVR in patients with HCV genotype 1 infection. Phase I data of patients (N = 7) with genotype 3 chronic hepatitis C treated with 400 mg of the drug BID for 4.5 days demonstrated that it was safe and tolerable and produced a maximum HCV viral load reduction of 3.68 log10 in six of seven patients who achieved an antiviral response.[57]
Phase II development of ACH-1625 began in the 3rd quarter of 2010 with the initiation of a randomized, double-blind, placebo-controlled trial evaluating the safety, tolerability and antiviral activity of QD ACH-1625 in combination with PR after 28 days of dosing (segment 1) and after 12 weeks of dosing (segment 2) in genotype 1 HCV-infected patients. Results from phase II studies of ACH-1625 have recently been released, and a preliminary report describes interim results of a 2-segment trial (N = 64). Patients who met the study criteria were randomized and stratified by HCV type and IL28B genotype in a 1:1 fashion to 48 weeks of PR plus ACH-1625 (200, 400 and 800 mg) or PBO administered concomitantly with the first 4 weeks of PR. HCV genotype-1a was the most common virus type (63%), whereas HCV genotype-1b infection was present in 25% of patients. After 4 weeks of ACH-1625 + PR, 81%, 75%, 76% and 20% of patients randomized to 200, 400, 600 mg or PBO had HCV RNA <25 IU/mL. The mean maximum decline in HCV RNA levels through the 4 weeks of combination ACH-1625 + PR therapy in the three escalating doses and PBO arms was 4.90, 4.63, 4.96 and 2.25 IU/mL log10, respectively. An interim analysis at 12 weeks of the first 35 patients enrolled demonstrated cEVR of 100% for each of the three doses. This potent antiviral activity was independent of the patients' IL28B status.[57] During the first 12 weeks of the ACH-1625 + PR therapy, there were no serious AEs or discontinuations resulting from safety/tolerability issues. Most reported AEs were transient, mild to moderate in severity and consistent with the tolerability/safety profile of PR. These results are notable, given that the participants had both viral and host factors that tend to reduce SVR.
Danoprevir
Danoprevir (RG7227, Roche and InterMune Pharmaceuticals, Brisbane, CA, USA) is another member of the second wave of first-generation PIs. The drug was evaluated in a randomized, placebo-controlled, 14-day multiple ascending-dose study in patients in four cohorts of treatment-naïve and one cohort of PR nonresponder patients with chronic HCV genotype 1 infection.[58] Treatment-naïve subjects were treated with one of four danoprevir dosing regimens (100 mg Q8h, 100 mg Q12h, 200 mg Q8h and 200 mg Q12h), whereas the nonresponders received 300 mg Q12h. The drug was safe and well tolerated; AEs were generally mild, transient and without association with treatment group or dose level. Maximal decreases in HCV RNA were −3.9 log10 and −3.2 log10 IU/mL in the treatment-naïve subjects receiving 200 mg Q8h and 200 mg Q12h, respectively. End-of-treatment viral decline in these two cohorts was within 0.1 log10 IU/mL of the viral load nadir. HCV RNA reduction in the nonresponders was more modest than that observed in upper dose of the naïve cohorts. The overall incidence of viral rebound was low (10/37) and was associated with R155K substitution in NS3 regardless of HCV subtype.
Danoprevir is being evaluated in treatment-naïve and nonresponder patients with chronic HCV genotype 1 infections with and without PegIFN or RBV, in combination with PR, and with RG7128 (nucleoside polymerase inhibitor). In the latter study, notably because interferon was not administered to any subject, 88 genotype-1 chronic hepatitis C patients were randomly assigned to seven treatment cohorts (n = 74) receiving various combinations of danoprevir (100 or 200 mg Q8h, or 600 mg or 900 mg BID), RG7128 (500 or 1000 mg BID) and PBO (n = 14) for up to 13 days.[59] Treatment-naïve patents received escalating doses, and prior nonresponders to PR were enrolled in higher-dose danoprevir cohorts. After the 14-day randomized study period, all participants started standard therapy with PR. The primary outcome was changes in HCV RNA viral load from baseline to day 14 in patients who received 13 days of combination therapy. All participants who completed treatment were included in the analysis. The mean decreases in HCV RNA from baseline to day 14 ranged from 3.7 to 5.2 log10 IU/mL in the danoprevir/polymerase inhibitor group. Subjects who received the highest doses of both drugs had a median decrease in viral load of 5.1 log10 IU/mL at day 14. Responses were similar in genotype-1a and genotype-1b infections. The combination was well tolerated and offers the potential of a PegIFN-free treatment combination. In addition, low-dose ritonavir, to strongly inhibit CYP3A, has been used to boost trough concentrations of danoprevir and potentially decrease the drug's overall toxicity.
Vaniprevir
Vaniprevir (MK-7009, Merck & Co., Inc.) is a NS3/4A PI that has demonstrated significant preclinical activity against HCV genotypes 1 and 2.[60] A randomized, placebo-controlled, double-blind 28-day study of the drug (600, 800, 1200 mg) + PR vs PR alone was conducted in treatment-naïve patients with chronic genotype-1 HCV infection.[61] Combination PR was administered for the full 48 weeks in all subjects. The primary endpoint was the per cent of subjects with viral suppression below the lower limit of detection at day 28 (RVR). In the ITT analysis, 71–83% of subjects achieved RVR vs only 5% in the PR-only treatment arm (P < 0.001). Virologic failure occurred in both 300 mg BID and 800 mg QD groups, with emergence of resistant strains. There were no serious AEs, and the most common AEs occurred at similar rates across all groups including PBO and were headache, nausea, fatigue and influenza-like symptoms. Investigators are currently recruiting subjects for a 48-week trial of vaniprevir + PR from those who have previously received the drug. The clinical development of this compound is on hold because of competing compounds within the Merck pipeline.
MK-5172
MK-5172 (Merck & Co., Inc) appears to be a second-generation PI with pan-genotypic activity. In patients with genotype-1 or genotype-3 chronic hepatitis C (N = 12), a once-daily dose of 400 mg for 7 days was generally well tolerated.[62] Mean maximum reductions from baseline of HCV RNA were 5.4 and 3.98 log10 for genotypes 1 and 3, respectively. Five of six genotype-1 patients had HCV RNA levels below the detectable limit during treatment. The mean time to nadir was more than 2 days after the last dose; plasma HCV RNA levels had returned to baseline by at the time of the 1-month follow-up visit. Additional studies are planned.
ACH-2684
ACH-2684 (Achillion Pharmaceuticals) also appears to be a second-generation PI. It shows highly potent pan-genotypic inhibition of HCV replication in vitro (genotypes 1-6). Preliminary evidence has shown that ACH-2684 exhibits IC50 values against NS3 proteases from genotypes 1–6 ranging from 0.04 to 0.7 nm. ACH-2684 retained its potency when tested against known resistant mutants including NS3 proteases carrying mutations at R155, A156 and D168 (IC50 0.2–2.2 nm). Furthermore, ACH-2684 does not inhibit CYP enzymes or activate CYP transcription; it is metabolically stable in human liver microsomes and shows high liver distribution in preclinical animal species.[63] ACH-2684 is currently undergoing phase I clinical studies.
NS5A Inhibitors
Daclatasvir Daclatasvir (BMS-790052, Bristol-Myers Squibb) is a potent, oral NS5A inhibitor. Doses of 1–200 mg were shown to be safe and tolerable in a phase I single ascending-dose study of healthy, non-HCV-infected subjects.[64] In a double-blind, placebo-controlled trial of single doses of the drug (1, 10, 100 mg) to patients with HCV genotype-1 infection (N = 18), the maximum dose produced a mean maximum reduction in HCV RNA of −3.6 log10 IU/mL, an effect that was maintained for 144 h.
The drug was studied in combination with the PI asunaprevir (BMS-650032) in a randomized, open-label phase IIa study of noncirrhotic patients with HCV genotype-1 infections who were nonresponders to PR (N = 21), a population whose viral infection is notoriously difficult to clear. The trial design compares the NS5A inhibitor plus the PI (n = 11) together and in combination with PR (n = 10) for 24 weeks. Double and triple combination therapy produced SVR 12 results of 36% and 100%, respectively.[49] At 24 weeks, double and triple combination therapy produced SVR in 36% and 90% of patients, respectively. The synergistic properties of the two drugs were able to clear infection without concomitant PR in this subset of patients. These preliminary results suggest that 24 weeks of triple therapy has the potential to cure chronic hepatitis C in patients who are unable to achieve a 2 log10 or greater decrease in HCV RNA after 12 or more weeks of PR.
ACH-2928
ACH-2928 (Achillion Pharmaceuticals) has very high potency that is in the picomolar range. When studied against a panel of chimeric replicons carrying NS5A from genotype-1a and genotype-1b HCV-infected patients, average EC50 values were 13 and 1.9 pm for genotypes 1a and 1b, respectively. EC50 values for genotypes 2a, 4a and 5a replicons were <14 pm.[65] Potency against genotypes 6a and 3a chimeric replicons was 48 and 103 pm, respectively. ACH-2928 exhibits a good safety and pharmacokinetic profile that strongly supports once-daily dosing and is highly effective in combination with NS3 PIs, NS5B polymerase inhibitors, PegIFN and RBV. Phase I trials have now begun with this compound.
NS5B Nucleoside Polymerase Inhibitors
RG-7128 RG-7128 (Gilead, Foster City, CA, USA, and Roche Pharmaceuticals) is a prodrug of PSI-6130, an oral cytidine nucleoside analogue. Phase I studies have demonstrated that the drug is safe and tolerable. A phase IIb study (N = 408) evaluated the drug in 500-mg and 1000-mg doses for 8–12 weeks plus the combination of PR for a total of 48 weeks. Treatment with the NI for 8 or 12 weeks produced complete EVR in >80%vs <50% of controls treated with PR and was safe and well tolerated. In addition, there were no viral rebounds or resistance-related breakthroughs during the first 8 or 12 weeks of triple combination therapy. A second phase IIb study evaluated RG-7128 at a dose of 1000 mg BID for 24 weeks plus PR in treatment-naïve patients with HCV genotypes 1 and 4. Patients who achieve an RVR on the combination of the drug plus PR will discontinue all therapy at 24 weeks, a design that allows the efficacy of 24-week combination to be assessed in patients who usually receive PR for 48 weeks. In this design, SVR 12 weeks after discontinuation of the 24 weeks of treatment (SVR-12) is used as an endpoint. In a preliminary report, 24 weeks of RG-7128 plus PR for 24 weeks produced a very high rate of virologic suppression (91%) and an SVR-12 of 76%.[66]
PSI-7977
PSI-7977 (Gilead) is an investigational NS5B NI. In early 2011, investigators began screening treatment-naïve patients with genotype-1 infection for a phase IIb study in the United States evaluating the drug in combination with PR. The planned enrolment is ~300 patients infected with genotypes 1, 4, 5 or 6.[67] Patients will be randomized equally to one of three arms. Treatment arms are PSI-7977 400 mg QD for 12 or 24 weeks with PR for 48 weeks. The third 48-week arm will evaluate the 400-mg dose for 12 weeks, and then enrollees will continue the NI for another 12 weeks with or without RBV; patients will continue to receive PegIFN for the full 48 weeks. The primary endpoint of the trial will be the safety and tolerability of PSI-7977 400 mg QD in combination with PR over 12 or 24 weeks. In a study of genotype-1, treatment-naïve, noncirrhotic subjects with chronic hepatitis C, treatment with PSI-7977 plus PR produced rapid reductions in HCV RNA as early as day 3 and in 98% of patients vs 19% of controls by week 4.[68] In addition, data have recently been reported from 24 treatment-naïve patients with HCV genotype 2 (n = 15) or 3 (n = 9) receiving 400 mg PSI-7977 QD in combination with PR for 12 weeks. All 24 patients achieved RVR (HCV RNA <15 IU/mL) after 4 weeks of treatment, with no differences in treatment efficacy according to either IL28B genotype or HCV genotype. Safety laboratory changes and adverse events were similar to clinical experience with PR, and no patients discontinued treatment because of adverse events.[69]
The antiviral profile of PSI-7977 led investigators to conduct a small, open-label, 12-week trial of interferon-free treatment with the NS5B NI.[70] Forty patients with chronic hepatitis C received 400 mg of PSI-7977 QD plus RBV; they were then randomized to PegIFN for 4, 8, 12 weeks or no interferon. By 4 weeks, all patients had HCV suppression to <15 IU/mL, regardless of the duration of interferon therapy. The PegIFN-free regimen was well tolerated and not associated with differences in viral kinetics, virologic breakthrough or failure to normalize ALT. Serious adverse events were 32% more common in patients treated with PegIFN. While RBV therapy was still accompanied by anaemia, haemoglobin levels were higher than in patients who also received the interferon. The manufacturer has now announced plans for a phase III, 12-week, all-oral, interferon-free regimen for patients with chronic hepatitis C, regardless of viral genotype or ability to take interferon therapy.[71]
INX-189
INX-189 (Inhibitex) is a phosphoramidate analogue of the nucleoside 2′-C-methylguanosine designed to deliver the monophosphate form of the nucleotide intracellularly, bypass the first rate-limiting phosphorylation and facilitate rapid conversion to the active triphosphate. Both in vivo and in vitro studies have demonstrated that INX-189 is rapidly metabolized to the therapeutically active triphosphate.[72,73] In addition, studies of INX-189-resistant HCV replicons identified 2 NS5B mutations that may be selected in the clinical setting. However, these mutations result in a dramatic loss of viral fitness and appear to increase the potency of RBV, suggesting a low likelihood for the generation of escape mutants with the clinical use of INX-189.[74] A preliminary report of phase Ib trial data for INX-189 indicated potent and dose-dependent antiviral activity. In this double-blind, placebo-controlled, dose-escalation study, 70 treatment-naïve HCV genotype 1 patients were randomized to one of seven dosing cohorts (five monotherapy, two combination therapy with RBV). Once-daily dosing with 9, 25, 50 or 100 mg for 7 days produced median HCV RNA reductions of −0.64, −1.00, −1.47 and −2.53 log10 IU/mL, respectively. One dose of 50 mg followed by once-daily dosing with 9 mg for 6 days produced a median HCV RNA reduction of −0.50 log10 IU/mL. When used in combination with RBV for 7 days, once-daily 9 mg INX-189 and 25 mg INX-189 produced a median HCV RNA reduction of −0.75 and −1.56 log10 IU/mL, respectively.[75]
NS5B Non-nucleoside Polymerase Inhibitors
Filibuvir Filibuvir (PF-00868554; Pfizer Inc., New York, NY, USA) is a NNI of NS5B. The antiviral characteristics, pharmacokinetics, and safety and tolerability of multiple doses of filibuvir have been evaluated in both treatment-naïve and treatment-experienced patients with HCV genotype-1 infections in 2 phase Ib clinical studies.[76] The first trial was a randomized, placebo-controlled dose-escalation study, whereas the second was a nonrandomized, open-label trial. Doses ranged from 200 to 1400 mg for 3–7 days. Genotypic changes in the NS5B nucleotide sequence following short-term filibuvir therapy were also assessed. The drug inhibited viral replication in a dose-dependent manner. In treatment-naïve patients, the maximum HCV RNA change from baseline ranged from −0.97 log10 IU/mL (100 mg BID) to −2.30 log10 IU/mL (700 mg BID). In treatment-experienced patients, an HCV RNA reduction of 2.20 log10 IU/mL was achieved with filibuvir 450 mg BID. The NNI was well tolerated in both studies, and adverse events were mild or moderate in severity. No discontinuations, serious AEs or deaths were reported. In the mutational analysis, sequencing of NS5B identified residue 423 as the predominant site of mutation following treatment with filibuvir. The phase II FITNESS trial is currently enrolling treatment-naïve, genotype-1 patients to receive the NNI (300, 600 mg) + PR for 24 weeks with or without an additional 24 weeks of PR. However, the clinical development of this agent has been placed on hold.[77]
VCH-222
VCH-222 (Vertex Pharmaceuticals) is an oral NNI of NS5B polymerase whose viral dynamics have been evaluated recently. In a study of treatment-naïve genotype-1a and genotype-1b HCV-infected patients (N = 5), 750 mg BID administered for 3 days produced a 3.7 log10 decrease in median HCV RNA levels.[78] The results were consistent between patients and across genotype 1 subtypes. In clinical evaluations to date, VCH-222 has been well tolerated, with no serious AEs observed. VCH-222 has completed 28-day nonclinical toxicology studies in two species. In a phase 2 trial, treatment-naïve patients with genotype 1 chronic hepatitis C (N = 106) were randomized to one of four arms: VCH-222 (100 or 400 mg) plus telaprevir (1125 mg) (V100 + T, V400 + T), V100 + T+PR and V400 + T+PR. HCV RNA levels, safety and tolerability were assessed at regular intervals. Both of the V + T arms were discontinued early (n = 47) because of viral breakthrough. In the ITT population, 57% (17/30) and 38% (11/29) of those in the V400 + T + PR and V100 + T + PR arms, respectively, were HCV RNA negative at week 2. In the high- and low-dose 4-drug arms, 90% (27/30) and 83% (24/29) of subjects were HCV RNA negative at week 12, respectively. Patients with undetectable HCV RNA at weeks 2, 8 and 12 were eligible to discontinue treatment after 3 months. In the V400 + T + PR arm 50% (15/30) of patients were able to stop all treatment at that time. The most frequently reported AEs were mild gastrointestinal intolerance or fatigue. On the basis of these results, an all-oral (V400 + T + R) 5th arm was opened for enrolment and a 6th arm may be added.
ABT-072
ABT-072 (Abbott, Abbott Park, IL, USA and Enanta Pharmaceuticals, Watertown, MA, USA) is a NNI inhibitor of the HCV NS5B polymerase. Phase I studies on safety and tolerability, and pharmacokinetics and pharmacodynamics have been conducted. In healthy volunteers, ABT-072 was well tolerated as both single and multiple doses. A phase II, open-label, 12-week pilot study to evaluate safety, tolerability, pharmacokinetics and pharmacodynamics of the drug in combination with RBV and the investigational oral PI ABT-450 is currently recruiting subjects. The primary outcome measure is the number of patients with undetectable HCV RNA at weeks 4 and 12. In a 12-week interim report, significantly greater numbers of patients treated with ABT-072 achieved EVR than those taking placebo + PR (P = 0.026).[79]
Setrobuvir
Setrobuvir (ANA-598; Anadys Pharmaceuticals, San Diego, CA, USA) is a DAA that inhibits activities of the HCV NS5B polymerase. In phase I studies, the drug has been demonstrated to exert potent antiviral activity and has a good safety profile. It is currently being studied in a phase IIb trial (N = 133) of treatment-naïve and treatment-experienced patients (incomplete responders, relapsers and nonresponders) infected with HCV genotype 1.[80] The treatment regimen is the NNI (200 mg BID) in combination with PR. The trial is multicentre and multinational. The primary endpoint of the study is SVR at 24 weeks.
Cyclophilin Inhibitors
Alisporivir Alisporivir (Debio-025; Debiopharm Group, Lausanne, Switzerland) is a nonimmunosuppressive form of cyclosporin. In cell cultures, the drug exhibits potent activity against both HCV and HIV. In HCV-infected cells, alisporivir's activity is mediated through cyclophilin B, a host protein that NS5B requires for maximum RNA binding. In a phase I trial of patients with HCV/HIV coinfection, 2 weeks of monotherapy produced a mean decline in HCV RNA of −3.6 log10.[81] In a phase II dose-ranging study of HCV-infected patients who were treatment naïve, the combination of the cyclophilin B inhibitor and PegIFN produced an additive antiviral effect. Combination of the PegIFN plus 600 or 1000 mg of alisporivir produced mean week 4 declines in HCV RNA of −4.61 log10 IU/mL and −4.75 log10 IU/mL, respectively.[82] In an interim analysis of a dose-ranging study of the drug in HCV genotype-1 patients who were nonresponders to PR (N = 50), the cyclophilin B inhibitor at 400 mg QD with or without PR did not significantly reduce viral load. However, a significant reduction in HCV RNA was produced with combination PR therapy plus either the 800 mg dose QD or when it was administered as a loading dose of 400 mg BID for 7 days and then 400 mg QD. A preliminary report from the manufacturer indicates that treatment with alisporivir plus PR for 48 weeks produced a SVR of 76% in genotype-1, treatment-naïve patients.[83]
Future HCV Treatment and Changing Standard of Care Characteristics of Emerging Therapies
Evidence from current trials suggests that PR will remain a central element in many future HCV regimens. Lessons from HIV over the last two decades, and with the development of highly active antiretroviral therapy (HAART), suggest that the treatment for HCV will be based upon 'cocktails' of drugs to avoid viral escape, prevent resistance, treat resistant strains and improve safety and tolerability. At the present time, the components of the ideal regimen are not known. Concerns about the use of multiple drugs mirror those identified in the HIV-infected population: pill burden, tolerability, safety and adherence.[84] A cautionary note is that resistance to some DAA can develop within days and persist for years.[85] Thus, it is important to minimize exposure to the various drugs during early clinical trials to afford participants an opportunity to attain SVR when they are offered potentially curative combination regimens.
Human Genomics
As with virologic and individual host factors, there is evidence that patient-based genomic studies may help identify patients who require more individualized treatment. More than 3.3 billion base pairs are present in the human genome; excess of 10 million of them may be single-nucleotide polymorphisms (SNPs). Already a number of SNPs have been identified that are associated with response to chronic hepatitis C treatment.[86] Because of linkage disequilibrium, however, they may all not be independent predictors. One clinically significant association is between a SNP on chromosome 19 within the region of the IL28B gene.[87] This single SNP affects viral kinetics and is highly correlated with a patient's response to PR therapy. It is reasonable to believe that this is a marker of 1 aspect of the immune system's multiple interactions with the virus.
A major limitation of RBV is drug-associated anaemia; it may contribute to interferon-associated fatigue, and in some patients, RBV haemolysis is so severe that the drug must be discontinued. Haemolysis appears to be the result of depletion of red blood cell (RBC) free phosphate as RBV is phosphorylated to RTP. As the intracellular pool of free phosphate declines, less ATP can be generated and the erythrocytes' membrane cannot be protected from oxidative injury. Although inosine triphosphate pyrophosphate (ITP) cannot be used directly by human RBC ATPase, it can substitute for GTP to regenerate ATP from AMP.[88] The presence of the SNPs in the ITP pyrophosphatase gene has been demonstrated to produce a dose-related decrease in the risk of RBV haemolysis.[89] Compared with wild-type ITPase, patients with polymorphisms have higher RBC ITP levels that can be used to restore ATP and maintain erythrocyte membrane integrity. ITPase activity and other markers may eventually be integrated into practice along with other host and viral factors to identify candidates for specific combinations of PR and DAA.
Current and Future Challenges in HCV Treatment
We are in an exciting era in which potential therapies for chronic hepatitis C are about to dramatically increase in number. However, as shown in the table, there are a number of potential challenges to improving patient outcomes (Table 2).
Table 2. Potential challenges to future treatment of patients with hepatitis C virus (HCV)
| Should pegylated interferon plus ribavirin remain a central element of treatment? |
| Should treatment be deferred until the ideal combination of drugs is available (i.e. warehousing)? |
| How should patients be stratified for treatment, and what is the role of genomics? |
| What is the optimum duration of treatment? |
| Should population-based screening measures be increased? |
| Who is going to treat the large numbers of patients who will be candidates for the newer combinations? |
| How will we establish the infrastructure to manage and follow the large numbers of patients with chronic hepatitis C? |
| How will the healthcare system handle the economic burden of highly active anti-HCV therapy when large numbers are being treated? |
| Will reimbursement be sufficient to compensate healthcare providers for the efforts necessary to manage and follow patients while they are being treated? |
| As the potential for sustained virologic response (SVR) increases dramatically, will patients be willing to tolerate a potential increased number of side effects to achieve better outcomes? |
Answers to these challenges are in various stages of discovery. Clinical trials continue to test different DAA with and without PR. Interferon-free regimens are also being examined. Resistance issues will become clearer as different drugs and combinations are evaluated. It may be that the hepatologists and infectious disease specialists will be overwhelmed with patients requesting treatment once optimum therapy is identified. As a result, community-based practitioners may begin to treat patients with uncomplicated chronic hepatitis C, while hepatologists and infectious disease specialists will continue to treat patients with comorbidities and HIV/HCV-coinfected patients, respectively.
Conclusions
As our understanding of HCV molecular biology has advanced, investigators have been able to develop designer drugs that target specific steps in the infection and replication of HCV. The introduction of DAA and creation of highly active anti-HCV therapeutic drug regimens has the potential to produce a dramatic improvement in outcomes in patients with chronic hepatitis C. However, the future also has the potential to challenge the healthcare system's ability to deliver the promise of these new medications.
http://www.medscape.com/viewarticle/766317
No comments:
Post a Comment