Correlation Between microRNA Expression Levels and Clinical Parameters Associated With Chronic Hepatitis C Viral Infection in Humans
Rebecca T Marquez; Sarmistha Bandyopadhyay; Erik B Wendlandt; Kathy Keck; Brandon A Hoffer; Michael S Icardi; Randolph N Christensen; Warren N Schmidt; Anton P McCaffrey
Authors and Disclosures
© 2010 Nature Publishing Group
Abstract and Introduction
Materials and Methods Results Discussion References
Abstract
MicroRNAs (miRNAs) are small RNAs that regulate gene expression pathways. Previous studies have shown interactions between hepatitis C virus (HCV) and host miRNAs. We measured miR-122 and miR-21 levels in HCV-infected human liver biopsies relative to uninfected human livers and correlated these with clinical patient data. miR-122 is required for HCV replication in vitro, and miR-21 is involved in cellular proliferation and tumorigenesis. We found that miR-21 expression correlated with viral load, fibrosis and serum liver transaminase levels. miR-122 expression inversely correlated with fibrosis, liver transaminase levels and patient age. miR-21 was induced ~twofold, and miR-122 was downregulated on infection of cultured cells with the HCV J6/JFH infectious clone, thus establishing a link to HCV. To further examine the relationship between fibrosis and the levels of miR-21 and miR-122, we measured their expression levels in a mouse carbon tetrachloride fibrosis model. As in the HCV-infected patient samples, fibrotic stage positively correlated with miR-21 and negatively correlated with miR-122 levels. Transforming growth factor β (TGF-β) is a critical mediator of fibrogenesis. We identified SMAD7 as a novel miR-21 target. SMAD7 is a negative regulator of TGF-β signaling, and its expression is induced by TGF-β. To confirm the relationship between miR-21 and the TGF-β signaling pathway, we measured the effect of miR-21 on a TGF-β-responsive reporter. We found that miR-21 enhanced TGF-β signaling, further supporting a relationship between miR-21 and fibrosis. We suggest a model in which miR-21 targeting of SMAD7 could increase TGF-β signaling, leading to increased fibrogenesis.
Introduction
Hepatitis C virus (HCV) is a positive strand RNA virus that infects 170 million people worldwide. Chronic HCV infection causes normally quiescent hepatocytes to divide repeatedly, leading to fibrosis, cirrhosis and occasionally progression to hepatocellular carcinoma (HCC). Fibrosis is characterized by excessive deposition of extracellular matrix.[1] In situ detection of HCV RNA in human liver suggests that liver injury is, at least in part, a function of HCV replication in hepatocytes.[2, 3]
microRNAs (miRNAs) are small non-coding RNAs that inhibit messenger RNAs (mRNAs) by binding to their 3′ untranslated regions (UTRs). miRNAs are predicted to repress up to 1/2 of all human genes post-transcriptionally through translational arrest and/or mRNA degradation.[4] miRNAs regulate diverse functions, including cell proliferation and apoptosis, and they are dysregulated in cancers, including HCC.[5–10]
Several studies have shown a relationship between HCV and host miRNAs, especially miR-122, the most abundant miRNA in the liver. Knockdown of the miRNA processing machinery in cultured cells significantly reduced HCV viral RNA.[11] In cultured cells, miR-122 dramatically enhanced,[12] but was not absolutely required for, HCV replication.[13] Furthermore, miR-122 binds to BACH1, which regulates HCV via heme oxygenase 1,[14] providing additional evidence of the relationship between miR-122 and HCV.
The relationship between HCV infection, miR-122 and the interferon (IFN) response is incompletely understood. Pederson et al [15] showed that IFN treatment of cells reduced miR-122 levels and elevated the levels of six other miRNAs. Inhibition of miR-122 and overexpression of these six miRNAs recapitulated IFN-mediated inhibition of HCV replication in cultured cells. Sarasin-Filipowicz et al [16] measured miR-122 levels in patients before and after IFN treatment. Surprisingly, treatment non-responders had significantly decreased pretreatment levels of miR-122, although miR-122 enhances replication in culture. After treatment, miR-122 levels remained unchanged in spite of the previous results showing that miR-122 was downregulated by IFN in culture. These contrasting results highlight the importance of complementing in vitro studies with experiments using samples from chronically infected HCV patients.
miRNAs could be novel diagnostic/prognostic markers and new therapeutic targets. Compared with mRNA expression profiles, data on miRNA expression during tumorigenesis were more accurate in tumor typing[17] and correlated with clinical outcomes.[18] Antisense oligonucleotides significantly reduced the levels of miR-122 in non-human primates[19] and thus could represent a new approach for HCV treatment. Clearly, there are important clinical implications to identifying miRNAs involved in HCV infection.
Four studies have examined miRNA levels in HCC samples with or without previous HCV infection.[5–7,20] Identifying HCV-specific effects in these studies is complicated by the overall dysregulation of miRNAs observed in tumors.
In this study, we analyzed the expression levels of miR-21 and miR-122 in needle biopsies from non-infected and HCV-infected patients. miR-21 was chosen because of its implication in control of proliferation and tumorigenesis.[10,18,21–25] Interestingly, miR-21 and miR-122 expression levels correlated with histological evidence of HCV liver disease and with stages of fibrosis in a carbon tetrachloride (CCl4) mouse model of liver fibrosis. We also showed that miR-21 represses a luciferase reporter containing the 3′UTR of SMAD7, a negative regulator of transforming growth factor β (TGF-β) signaling. Furthermore, miR-21 overexpression in hepatoma cells leads to increased luciferase production of a TGF-β reporter, confirming a positive relationship between miR-21 and TGF-β signaling, possibly through SMAD7. These results provide novel insights into the interaction between HCV, fibrosis and miRNAs.
Patients and Clinical Samples
Patient clinical data are listed in Table 1 . This study was approved by the University of Iowa Institutional Review Board, conformed to the human experimentation guidelines of the US Department of Health and Human Services, and written informed consent for specimen use was obtained. Uninfected control liver samples (n=4) were obtained from the surgical margins of benign liver masses that were histologically and pathologically normal.[26] HCV-positive samples obtained during routine patient work-ups were stained with hematoxylin and eosin (H & E) or Masson's trichrome and scored blindly for fibrosis and necroinflammatory activity using a METAVIR scoring system[27] as described previously.[28] Patients studied were designated HCV-positive if their serum tested positive in an enzyme-linked immunosorbent assay (EIA.3) for HCV antibodies. HCV-positive patients had active HCV viremia within 3 months of biopsy as determined by commercial reverse transcription polymerase chain reaction (RT-PCR) assay (Monitor Assays; Roche Diagnostic Systems, Branchburg, NJ, USA). HCV genotypes were determined by second generation tests (INNO-LiPA, Innogenetics, Zwijnaarde, Belgium). All HCV-positive patients had characteristic histological findings on liver biopsy.[29] Other causes of chronic liver disease were ruled out as described previously.[30] Histopathological determinations used routine 2.0–2.5 cm percutaneous core liver biopsy samples. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) measurements are reported in international units/ml, and reference ranges were 0–37 and 0–35, respectively. Viral load is reported in international units/ml.
RNA Isolation
Total RNA was isolated from 2 mm sections of snap-frozen core liver needle biopsies stored at −80 °C and processed using Trizol Reagent (Invitrogen, Carlsbad, CA, USA). RNA integrity was measured by an Agilent 2100 Bioanalyzer and a RNA 6000 Nano Chip Kit (Agilent Technologies, Santa Clara, CA, USA).
COL1A1, MX1 and HCV RNA Quantitative (Q)-RT-PCR
cDNA was synthesized from total RNA using the High Capacity Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) and quantitative real-time PCR was performed using 2 × Sybr Green Master Mix (Applied Biosystems). The following primers were used: COL1A1: sense 5′-GTTTGGAGAGAGCATGACCGA-3′, antisense 5′-TGGACATTAGGCGCAGGAA-3′; MX1: sense 5′-CACGAGTTCCACAAATGGAGTA-3′, antisense 5′- TTCACGATTGTCTCAAATGTCC-3′; HCV: sense 5′-CGGGAGAGCCATAGTGG-3′, antisense 5′-AGTACCACAAGGCCTTTCG-3′ as described previously.[31] mRNA and HCV expression values were normalized to glyceraldehyde 3-phosphate dehydrogenase. For miRNA measurements, three independent cDNAs were synthesized from 15 ng total RNA using the TaqMan MicroRNA Assay protocol (Applied Biosystems), and Q-PCR levels were measured using the TaqMan® Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems). All miRNA expression values were calculated from a standard curve and normalized to an endogenous control, either RNU6B or RNU48.
CCL4 Mouse Fibrosis Model
Eight-week-old Balb/c mice (National Cancer Institute, Bethesda, MD, USA) were injected intraperitoneally with 0, 7.5, 10 or 15 μl CCl4 mixed with 100–250 μl mineral oil, twice a week for 4 or 7 weeks. We chose these doses to obtain varying degrees of fibrosis. Four days after the last CCl4 injection, mice were killed, and RNA was prepared. Paraffin sections were stained with Masson's trichrome or H & E by the University of Iowa Central Pathology Core. A board certified pathologist (MSI) staged the slides in a blinded manner. As CCl4-induced fibrosis in mice does not mirror the fibrosis observed in humans exactly, it was not appropriate to use a traditional staging scheme such as the Batts–Ludwig criteria.[32] We therefore made the following modifications. Stage 0=no significant fibrosis or necrosis. Stage 1=mild increase in fibrosis around the central veins and occasionally around the portal tracks. Minimal-to-no hepatocyte necrosis. Stage 2=mild-to-moderate increase in fibrosis around the central veins and occasionally around portal areas with thin incomplete bridging between the central and central areas. Mild hepatocyte necrosis primarily in the central regions. Stage 3=mild-to-moderate fibrosis around the central veins with complete bridging between the central and central areas; this was accompanied by moderate central and bridging necrosis.
Statistical Analysis
The non-parametric Spearman's rank correlation test was used to determine the correlation between miRNA expression levels and the following clinical parameters: fibrosis, viral load, AST and ALT serum levels, genotype and age; this test was also used to correlate miR-21 levels with fibrotic stage and collagen mRNA levels in the CCl4 fibrosis model. An unpaired two-tailed Student's t-test was used to compare miR-21 or miR-122 levels in uninfected and HCV cell culture infectious clone (HCVcc)-infected Huh7.5 cells. Correlations with P-values ≤0.05 were considered significant and are indicated on graphs with an asterisk.
SMAD7 3′ UTR Luciferase Reporter Constructs
We amplified the 3′UTR of SMAD7 (NM_005904, nucleotides 1569–3094) from human liver cDNA and cloned it into the XhoI/NotI sites downstream of the Renilla luciferase gene in the psiCHECK-2 vector (Promega, Madison, WI, USA). The miR-21 seed in the SMAD7 3′UTR was deleted using the QuickChange XL Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA).
Cell Culture, Transfection and Luciferase Reporter Assays
HEK-293 and Huh7.5 cells were grown in Dulbecco's Modified Eagle Medium/10% fetal bovine serum/1 × glutamine/1 × pyruvate/1 × non-essential amino acids/1 × penicillin/1 × streptomycin (Invitrogen). For SMAD7 luciferase reporter assays, HEK-293 cells were plated at a density of 4 × 105 cells in 24-well plates. Cells were co-transfected with 50 ng of psiCHECK-2-SMAD7 (wild-type or miR-21 seed sequence-deleted) with 50 nM miR-21 mimic (Ambion, Austin, TX, USA) or negative control mimic (Ambion) using Lipofectamine 2000 Transfection Reagent (Invitrogen). Cells were harvested 24 h later and assayed using the Dual-Luciferase Reporter Assay System (Promega) using a MicroLumatPlus luminometer (Berthold Technologies, Oak Ridge, TN, USA). Renilla luciferase values were normalized to firefly luciferase values expressed from the same psiCHECK-2 vector. For the TGF-β luciferase reporter assays, Huh7 cells were plated at a density of 1.5 × 105 in 24-well plates. Cells were co-transfected with 250 ng of p3TP-Lux vector, 0.05 ng Renilla vector as a normalizing control and either 50 nM miR-21 mimic (Ambion) or negative control mimic (Ambion) using Lipofectamine 2000 Transfection Reagent (Invitrogen). After 8 h, media was replaced with complete media containing 0.2% serum and incubated overnight. Cells were treated with 200 pM TGF-β (R and D systems, Minneapolis, MN, USA), harvested 8 h later and assayed using the Dual-Luciferase Reporter Assay System (Promega) using a MicroLumatPlus luminometer (Berthold Technologies).
HCV Infectious Clone-infected Cells
Huh7.5 cells and the J6/JFH1 HCVcc were generous gifts from Dr Charles Rice (Rockefeller University, New York, NY) and were prepared and maintained as described.[33] Huh-7.5 cells were infected with HCVcc at a multiplicity of infection of 2.2. At 3, 4 and 6 days after infection, RNA was harvested, and HCV Q-RT-PCR was performed. Day 6 samples were trypsinized at day 4, and one-third of the cells were re-plated and cultured until day 6. Total RNA was harvested, and HCV Q-RT-PCR was performed.
Isolation of Primary Rat Hepatic Stellate Cells
Rat hepatic stellate cells (HSC) were isolated and cultured as previously described.[34] Total RNA was isolated from quiescent HSC immediately after isolation (day 0) or from activated HSC maintained in culture for varying lengths of time using the TRIzol reagent (Invitrogen).
miRNA Expression is Altered During HCV Infection in Human Livers
We compared the expression of miR-21 and miR-122 in HCV-infected and uninfected human livers. The HCV-infected patient samples represented varying stages of fibrosis ( Table 1 ) while maintaining low inflammatory grades. This minimized the measurement of miRNAs expressed from infiltrating lymphocytes that may confound results. Both miRNAs showed aberrant expression in HCV-infected livers (Figure 1).

Figure 1.
miR-21 and miR-122 expression were dysregulated in HCV-infected human liver samples. Data are presented as ratio of miRNA expression in HCV-infected human liver relative to mean expression in uninfected liver (N=4) represented by the black bar at left. Patient samples are arranged by increasing fibrosis stages (0–4) indicated at bottom of graph. Q-RT-PCR was performed in triplicate and error bars represent ± standard error of the mean (s.e.m.).
Altered miRNA Expression Correlates With Clinical Parameters
Table 1 shows gender, age, genotype, serum AST levels, ALT levels, peripheral viral load and fibrotic stage for patients tested. Spearman's rank correlations with miRNA expression levels are shown in Table 2 . miR-21 expression correlated with fibrotic stage, viral load and AST and ALT serum levels but not genotype or age. In contrast, miR-122 expression inversely correlated with fibrotic stage as well as AST and ALT serum levels; thus, miR-122 expression decreased concomitantly with biochemical evidence of hepatocyte damage. As it was shown previously that miR-122 levels decreased in cultured hepatocytes on treatment with type I IFNs,[15] we measured the levels of the IFN-responsive gene, MX1, in our patient samples. We observed a strong inverse correlation between miR-122 levels and MX1 (r=−0.653, P=0.0013, Table 2 ) and a positive correlation between miR-21 and MX1 levels (r=0.431, P=0.050, Table 2 ). Thus, we show that miRNA levels correlate with various clinical parameters during chronic HCV infection.
Potential Reasons for miR-21 Induction and miR-122 Repression
There are several possible explanations for miR-21 upregulation and miR-122 downregulation in high fibrotic HCV-infected patients. For example, HCV could alter the expression of miR-21 or miR-122. Alternatively, miR-21 could be upregulated or miR-122 could be downregulated during fibrogenesis. Below, we carried out experiments to distinguish among these possibilities.
miR-21 and miR-122 Expression Was Modulated in Cells Infected With an HCVcc Clone
Recently, Lindenbach et al [33] described a genotype 2a HCVcc that infects Huh7.5 cells, replicates and produces infectious virus particles. To determine whether HCVcc would alter the expression of miR-21 or miR-122, we infected Huh7.5 cells with HCVcc at a multiplicity of infection of 2.2. Cells were harvested at 3, 4 and 6 days, and immunocytochemical staining with HCV patient immune serum showed 80, 100 and 100% infection, respectively (data not shown). We observed statistically significant induction of miR-21 by HCVcc at 96 and 144 h (Figure 2a). miR-122 levels were significantly reduced on HCVcc infection at 144 h (Figure 2b). Thus, infection with HCV in culture causes the same alterations of miR-21 and miR-122 levels observed in chronically HCV-infected patients. However, this correlation was only observed in culture at later times after infection.

Figure 2.
Effect of HCVcc infection on levels of miR-21 and miR-122. Huh 7.5 cells were mock-infected or infected with HCVcc. miRNA levels were measured by Q-RT-PCR. (a) miR-21 levels relative to uninfected cells. (b) miR-122 levels relative to uninfected cells. Data are mean±s.e.m. from two independent experiments carried out in triplicate. * P<0.05.
miR-21 and miR-122 Expression Levels in a CCl4 Mouse Model of Liver Fibrosis
Expression levels of miR-21 positively correlated with fibrotic stage in chronically HCV-infected patients, while miR-122 levels were negatively correlated ( Table 2 ). To determine whether miR-21 or miR-122 overexpression is associated with fibrosis independent of HCV infection, we used the classical CCl4-induced mouse liver fibrosis model. Mice were treated with two different doses of CCl4 twice a week for 4 weeks to induce liver fibrosis. Four days after the final treatment, mice were killed. Paraffin sections were stained with Masson's trichrome, which stains for collagen. Although the CCl4 mouse fibrosis model is commonly used and experimentally tractable, it should be noted that fibrosis in this model resembles, but does not completely recapitulate, the fibrosis pattern observed in chronically infected HCV patients. We therefore modified the Batts–Ludwig criteria[32] as described in the Materials and methods section. A pathologist (MSI) assigned each section a fibrotic stage score in a blinded manner (representative Masson's trichrome stains, Figure 3a). Spearman's rank correlation showed that miR-21 levels from CCl4-treated mice significantly correlated with fibrotic stage (Figure 3b, r=0.359, P=0.029). In contrast, miR-122 levels inversely correlated with fibrotic stage (Figure 3C, r=−0.493, P=0.002). These results show that miR-21 levels are induced and miR-122 levels are repressed during fibrosis, independent of HCV infection.
Click Figure 3 To Enlarge:
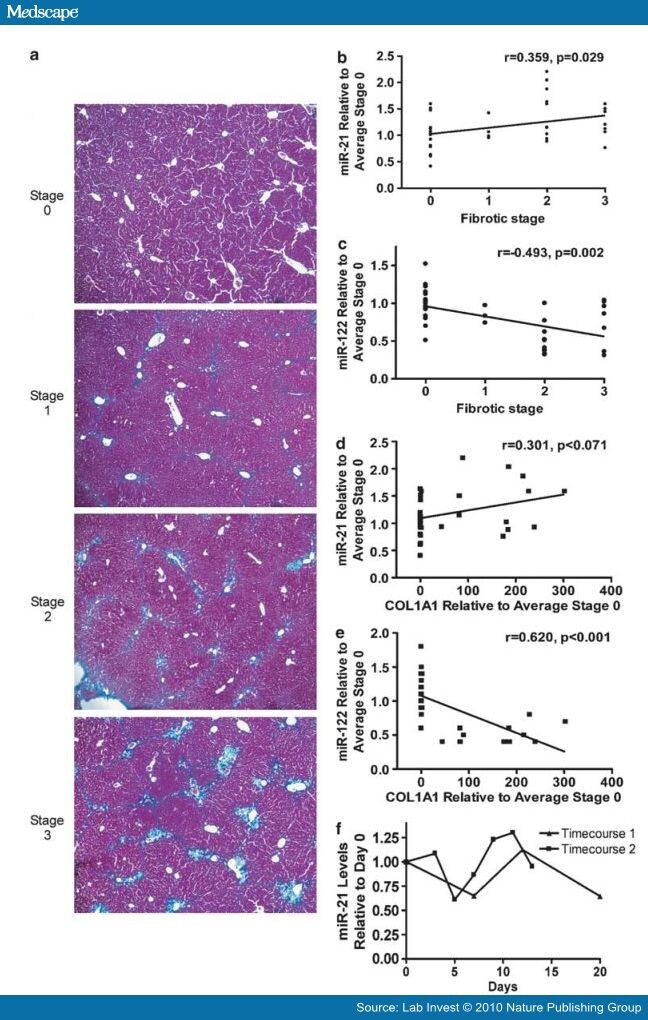
Figure 3.
CCl4-treated mice and activated primary stellate cells. (a) Stage of fibrosis is indicated (see Materials and Methods). Staging was carried out in a blinded manner. Representative images are shown. Magnification, × 10. Correlation plots are shown for (b) miR-21 expression vs fibrotic stage, (c) miR-122 expression vs fibrotic stage, (d) miR-21 expression vs COL1A1 expression and (e) miR-122 expression vs COL1A1 expression. In (b–e), miRNA and COL1A1 levels are expressed as fold change relative to the average miRNA or COL1A1 value of the 0 stage fibrosis group. (f) miR-21 expression relative to day 0 during a rat primary stellate cell activation timecourse.
A hallmark of fibrosis is the production of collagens. We used Q-RT-PCR to measure the level of COL1A1 mRNA in the livers of CCl4-treated mice. A Spearman's rank correlation analysis showed that COL1A1 levels were inversely correlated with miR-122 levels (Figure 3E, r=0.620, P<0.001). r="0.301,">

Figure 4.
miR-21 directly targets the 3′UTR of SMAD7. The predicted interaction between miR-21 and SMAD7 3′UTR. A mutant construct was created by changing nucleotides (in bold) to complementary bases. HEK-293 cells were co-transfected with luciferase reporter construct containing the 3′UTR of SMAD7 and miR-21 or negative control mimic. miR-21 overexpression inhibited the SMAD7 3′UTR luciferase reporter, while the mutant SMAD7 3′UTR reporter construct was not inhibited by miR-21 co-transfection. This showed the specificity of miR-21-mediated inhibition of SMAD7. Data are mean±s.e.m.; average of three independent experiments; * P<0.05.
miR-21 Overexpression Enhanced TGF-β Signaling
As miR-21 targets a negative regulator of TGF-β, we hypothesized that miR-21 overexpression might increase signaling in response to TGF-β. To test this, a TGF-β-responsive luciferase reporter and a miR-21 mimic (or negative control mimic) were co-transfected into Huh7 cells followed by treatment with TGF-β. Figure 5 shows that the miR-21 mimic, but not the negative control mimic, stimulated TGF-β signaling. These results further establish a relationship between miR-21 and the TGF-β signaling pathway.

Figure 5.
miR-21 overexpression increases TGF-β luciferase reporter signal in Huh7 cells. Huh7 cells were co-transfected with 250 ng p3TP-Lux, a TGF-β-responsive luciferase reporter construct, 0.05 ng Renilla normalization control vector and pre-miR-21 (50 nM) or negative control pre-miR (50 nM) to measure the effect of miR-21 on TGF-β signaling. Cells were treated with 200 pM TGF-β for 8 h to activate TGF-β signaling. Cells transfected with pre-miR-21 had a twofold increase in luciferase production as compared with negative control pre-miR. Relative light units are firefly/Renilla luciferase photons/s. Data are mean±s.e.m.; average of four independent experiments; * P<0.05.
Discussion
In this study, we identified two miRNAs that were dysregulated in HCV-infected patient samples. We found several correlations between miR-21 and miR-122 expression levels and clinical and histological findings. It is noteworthy that miR-21 levels correlated with viral load, fibrotic stage and serum liver transaminases (Table 2). miR-21 expression seems low at early fibrotic stages and increases with increasing stage of fibrosis. The correlation does not seem to be strictly linear. This may reflect the fact that accumulation of fibrosis may not only be related to the expression level of miR-21 but also to the duration of miR-21 upregulation during the course of HCV infection. This second variable is hard to account for since the time of HCV infection is almost never known precisely. Interestingly, infection of cultured hepatocytes with HCVcc at late times recapitulated the miR-21 upregulation observed in patients, suggesting that the virus or the host modulates miR-21 in response to long-term infection (Figure 2A). We also observed a correlation between miR-21 and fibrotic stage in the CCl4 mouse fibrosis model showing that fibrosis, independent of HCV, was also associated with miR-21 upregulation. Thus, multiple mechanisms may modulate miR-21 in the complex environment of the damaged, chronically HCV-infected human liver. miR-21 did not seem to be consistently upregulated during an in vitro activation time course of primary stellate cells, suggesting that the downregulation of miR-21 observed in HCV-infected patients was probably not due to reductions in miR-21 in this cellular compartment. From our study, we can not determine if miR-21 induction is causative for fibrosis. However, a previous study showed that miR-21 levels were elevated in damaged human hearts, and inhibition of miR-21 in mice reduced cardiac fibrosis during heart failure;[40] this raises the interesting hypothesis that miR-21 induction by HCV could result in liver fibrosis. Although miR-21 induction may be associated with HCV infection and fibrosis, several papers have shown miR-21 expression is associated with proliferation. miR-21 is upregulated in many cancers, including HCC,[10,21–24,41] and overexpression of miR-21 in HCC cell lines led to increased proliferation and migration. In addition, we recently showed that miR-21 was upregulated during the proliferative phases of liver regeneration in mice.[42] Therefore, miR-21 upregulation may also be due to increased proliferation of liver cells during HCV infection and/or during late stages of fibrosis.
Interestingly, miR-122 expression levels were inversely correlated with fibrotic stage, age and liver transaminases, but not viral load. miR-122 expression levels were most consistently downregulated at late stages of fibrosis. The inverse correlation between miR-122 and fibrosis is consistent with a previous report.[16] We also observed an inverse correlation between miR-122 levels and fibrotic stage in mice that was independent of HCV infection. From our study, it is not possible to determine if miR-122 downregulation is a cause or consequence of fibrosis. Antisense miR-122 inhibitors have been tested as potential HCV inhibitors in non-human primates.[43] Although our data suggest that inhibition of miR-122 correlates with fibrosis, long term miR-122 inhibition in African green monkeys did not result in liver pathology, suggesting that downregulation of miR-122 does not cause liver fibrosis.[19]
Our study revealed no correlation between miR-122 and viral load, but we did observe an inverse correlation between miR-122 and transaminase levels. These results are counterintuitive, because miR-122 greatly enhances HCV replication in cultured cells.[12] Thus, one might anticipate that decreased levels of miR-122 would reduce HCV replication, liver damage and lower serum transaminases. Recently, Sarasin-Filipowicz et al reported the surprising finding that IFN treatment non-responders had lower levels of miR-122 than did responders. These non-responders seemed to have pre-activation of IFN-responsive genes; this is consistent with our observation showing a strong inverse correlation between miR-122 and levels of the MX1 IFN-responsive gene in our patient samples.
TGF-β is critical in the development of liver fibrosis induced by a number of chemical and viral insults and TGF-β induces apoptosis in hepatocytes (reviewed in Meindl-Beinker and Dooley,[37] Schuster and Krieglstein[44]). SMAD7, which is upregulated by TGF-β, is a negative regulator of TGF-β signaling.[38] Overexpression of SMAD7 greatly ameliorated CCl4-induced fibrosis in mice.[37] In this study, we showed that miR-21 directly targeted SMAD7. The possibility that miR-21 upregulation during chronic HCV infection induces fibrosis by downregulating SMAD7 will require further study.
Several studies have shown a relationship between TGF-β signaling and miR-21. TGF-β stimulates processing of the primary miR-21 precursor (pri-miRNA) into mature miR-21.[45] Clinical studies correlated miR-21 and TGF-β expression in breast cancer patients.[46] A microarray study indicated that miR-21 may regulate, directly or indirectly, TGF-β2, bone morphogenetic protein 4 and epidermal growth factor.[47] These studies suggest that miR-21 forms negative feedback cycles regulating TGF-β signaling. In this study, we show for the first time that miR-21 represses a negative regulator of TGF-β signaling, SMAD7. This result raises the possibility that miR-21 may also form a positive feedback cycle mediated by SMAD7.
In summary, the correlation between miR-21 and viral load in patient samples, as well as its upregulation by HCVcc, suggests that HCV may upregulate miR-21. miR-21 is upregulated in both HCV-associated fibrotic patient samples as well as CCl4-mediated liver fibrosis/damage in the absence of HCV. The upregulation of miR-21 may be due to TGF-β processing of the miR-21 precursor into mature miR-21.[45] Upregulation of mature miR-21 would then lead to inhibition of SMAD7, which encodes a negative regulator of TGF-β signaling; this forms a positive feedback loop that may increase fibrosis. Our results highlight liver disease states that alter miR-21 levels and identify a novel miR-21 target that may further modulate those disease states.
Materials and Methods
Results
Discussion
References
No comments:
Post a Comment