{kind=link}
'
Received 22 July 2010; accepted 9 September 2010.
'
Agents that specifically target the replication cycle of the virus [specifically targeted antiviral therapies for hepatitis C (STAT-Cs)] by directly inhibiting the NS3/4A serine protease, the NS5B polymerase and NS5A are currently in clinical development. The need to achieve serum drug concentrations able to suppress viral replication is a key factor for a successful antiviral therapy and the prevention of resistance. Thus pharmacokinetics parameters became important issues for drugs used in the therapy of hepatitis C. The ratio of Cmin/IC50 (inhibitory quotient or IQ) can provide a surrogate measure of a drug's ability to suppress HCV replication, by taking into account the relationship between plasma drug levels and viral susceptibility to the drug. Ritonavir boosting may be a useful strategy to improve pharmacokinetic parameters. Characterising resistance to STAT-Cs in clinical trials is essential for the management of a drug regimen to reduce the development of resistance and thereby maximise SVR rate. The lesson of HIV therapy, provide a compelling case for the exploration of combinations of direct-acting antiviral agents.
Keywords: STAT-C, Pharmacokinetics, Ritonavir boosting, Resistance
Hepatitis C virus (HCV) chronically infects 180 million people worldwide with an estimated incidence of new cases of 3–4 million each year [1], [2]. In the developed world, HCV accounts for 50–76% of all primary liver cancer cases and 30% of all liver transplants, 1 and has been estimated to result in a reduction in overall life expectancy in infected individuals of between 8 and 12 years [3]. The current recommended treatment, consisting of a peginterferon plus ribavirin (Peg-IFN/RBV) combination achieves response rates ranging from 76% to 80% in patients infected with HCV genotypes 2 and 3 and from 40% to 50% in those with genotype 1 [3]. Although this kind of therapy will remain the Standard of Care (SOC) for the next years, more effective therapeutic options with shorter treatment durations are needed to increase the response rate in difficult to treat patients (mainly genotype 1) and reduce the impact of HCV infection and its associated complications.
So far, agents that specifically target the replication cycle of the virus [specifically targeted antiviral therapies for hepatitis C (STAT-Cs)] by directly inhibiting the NS3/4A serine protease (which processes the HCV polyprotein to generate mature viral proteins), the NS5B polymerase (which replicates the viral RNA genome) and NS5A (which functions as a part of the replicase complex) are currently in clinical development [4].
HCV is the only member of the hepacivirus genus of Flaviviridae. As with other Flaviviridae, the 9.4kb (+) sense RNA genome is translated into a single long polyprotein that is cleaved by both host signal peptidases and virally encoded proteases (NS2, NS3/4A) into 10 functional peptides (structural and nonstructural proteins). One of these proteins, the NS5B RNA-dependent RNA polymerase, catalyzes the direct copying of the viral genome into a replicative intermediate RNA. As there is no DNA intermediate (i.e. no reverse transcriptase activity), HCV is not known to be capable of a latent phase [5].

Download to PowerPoint
Fig. 1. HCV replication cycle and site of action of STAT-C.
2.2. Hepatitis C virus mutations, replicative fitness
As for human immunodeficiency virus (HIV) and hepatitis B virus (HBV), HCV- resistant variants are usually not as fit as wild type viruses, particularly if drugs to which they are resistant bind directly to the active site of a viral enzyme. However, a resistant variant can improve fitness by the accumulation of additional compensatory mutations.
The fitness of these HCV variants is typically estimated in vitro by measuring replication capacity (by transient replication in the replicon system) and enzymatic fitness (by measuring catalytic efficiency) [7], [8]. The resistant variants have varying degrees of decreased replication capacity. The NS3 A156T mutation, which confers resistance to many Protease Inhibitors (PIs), has significantly reduced NS3/4A catalytic efficiency and replication capacity [9], [10]. The NS5B nucleoside inhibitor-resistant mutation, S282T, also has decreased replication capacity [8]. The non-nucleoside inhibitor-resistant mutation P495A/L has decreased replication capacity, but fitness can be restored by compensatory mutations elsewhere in NS5B.
Recently, the in vivo fitness of viral variants with decreased sensitivity to the HCV PI telaprevir (TVR) has been estimated using a novel method that assessed growth rate in the absence of TVR selective pressure. The replicative fitness of different viral variants was inversely correlated with their degree of resistance to TVR [11]. Fitness of resistant variants is not only important in determining the probability of emerging resistance but also to predict if they will revert to wild type in the absence of drug selective pressure. Resistant variants with significantly impaired fitness will be replaced by wild-type viruses more rapidly in the absence of drug selective pressure [11].
2.3. STAT-C in clinical development
2.4. NS3/4a protease inhibitors
The HCV NS3 protein is a multifunctional protein that consists of an amino-terminal serine protease and a carboxy-terminal helicase/nucleoside triphosphatase domain [14] and is necessary for post-translational processing of the NS3–NS5 region of the HCV polyprotein to generate components of the viral RNA replication complex [14]. NS4a acts as a cofactor to facilitate the serine protease function. The helicase is thought to have a role in viral replication by unwinding the viral RNA [14]. The NS3/4a protease has been shown to be a key regulator of intracellular type I IFN pathways. Inhibitors of the NS3/4a protease therefore act to inhibit directly viral replication. We briefly reviewed the clinical characteristics of STAT-C agents, currently in phase of clinical development.
2.5. NS5B polymerase inhibitors
The HCV NS5B RNA-dependent RNA polymerase is a key enzyme involved in HCV replication, catalyzing the synthesis of the complementary minus-strand RNA and subsequent genomic plus-strand RNA from the minus-strand template. Both nucleos(t)ide and non-nucleos(t)ide polymerase inhibitors (NI/NNI) are currently in development. In addition, the replicative activity of the RdRp has recently been reported to be augmented by direct binding to cyclophilin B, a host cell isomerase [15]. A cyclophilin B inhibitor has also progressed to a phase 2 clinical development programme.
To describe the characteristics and the results obtained in clinical trial of each molecules is beyond the goal of this article which is focused on specific issues of the management.
A list of the drugs so far tested in clinical trials and the side effects of them are summarized in Table 1, Table 2.
Site of action and stage of development of molecules tested so far in clinical trials.
Life cycle step
Category....................Drug name........................Phase of development
HCV replication........Polymerase inhibitor........RG7128......Phase I
................................................................................VX-222.......Phase II
...............................................................................ABT-072.....Phase I
.............................................................................MK-3281......Phase I
...............................................................................PSI-7851.....Phase I
................................................................................ABT-333....Phase I
..................................................................................IDX184.....Phase II
................................................................................ANA598......Phase II
.................................................................................GS9190......Phase II
...................................................................................VX-759.....Phase II
...............................................................................PSI-7977......Phase IIa
...............................................NS5A inhibitor.........PPI-461.......Phase I
....................................................................................A-832......Phase II
.......................................................................BMS-790052......Phase II
................................................HCV polymerase..........VX-916.....Phase I
....................................HCV polymerase inhibitor......Filibuvir....Phase I
...............................................Cyclophilin inhibitor.....SCY-635....Phase I
.................................................................................Debio 025.....Phase II
........................................................NS4B inhibitor....Clemizole.....Phase I
......Post-translation processing...Protease inhibitor..RG7227....Phase II
.................................................................VX950 (telaprevir).......Phase III
.................................................................................IDX320.......Phase I
..............................................................................ACH-1625.......Phase I
...................................................................................VX-813.......Phase I
................................................................................PHX1766.......Phase I
..................................................................................GS-9256......Phase II
..............................................................................BI 201335.......Phase II
.........................................................SCH900518 (narlaprevir).......Phase II
.............................................................................TMC435......Phase IIa
................................................SCH 503034 (boceprevir).....Phase III
........................HCV protease inhibitor..............VX-500......Phase I
...................................................MK-7009 (vaniprevir)........Phase II
Table 2
Main side effects of STAT-C tested in clinical trials.
Class.................Drug..............Side effects
........................NS3/4a protease inhibitors......Boceprevir
Headache, rigor, myalgia and fever fatigue, anemia, nausea; adverse events were also the most commonly for PEG-IFNα2b monotherapy [46]
..............................................................................Telaprevir...Influenza-like illness, fatigue, headache, nausea, anemia, depression, and pruritus constipation, abdominal pain, gastroesophageal reflux disease, nausea, vomiting, headache, acute otitis media, and seasonal allergy. Decreases in hemoglobin, total white blood cell count, neutrophils, and platelets occurred during the study drug dosing period [13]
................................NS5B NIs......R1626....Vomiting and diarrhoea, moderate leokopenia; cytopenia, haematological toxicity, neutropenia, anemia, rash [13]
.......................................................R7128 4...............Haematological, toxicity, Headache, fatigue and chills [13]
.......................................................R7128 4.............Mild-moderate diarrhoea [13]
The need to achieve serum drug concentrations able to suppress viral replication is a key factor for a successful antiviral therapy and the prevention of resistance. Thus pharmacokinetics parameters became important issues for drugs used in the therapy of hepatitis C.
Although the HCV protease inhibitors may have a major clinical impact as a drug class, they have a relatively narrow therapeutic index because they require highly suppressive drug concentrations to prevent the emergence of resistance. The concept of protease inhibitor (PI) boosting was developed to overcome these issues [16].
When considering the pharmacokinetics of antivirals several parameters must be taken into account.
After multiple doses of a drug are administered over several days of treatment, the maximum and minimum concentrations of drug achieved after each dose reach constant levels.
Cmax=the peak or highest plasma concentration achieved during a dosing interval.
Tmax=the time taken to reach the highest observed plasma concentration.
Cmin=the lowest observed plasma concentration achieved during a dosing interval. Cmin is also called the ‘trough concentration’, and generally occurs at the end of the dosing interval

Download to PowerPoint
Fig. 2. Theoretical plasma concentration/time curve showing fundamental pharmacokinetics parameters.
3.1. Understanding parameters of antiviral efficacy
Inhibitory Concentration (IC)50/IC90 are the in vitro concentration of drug required to inhibit viral replication by 50%/90%.
IC50 and IC90 vary depending upon a number of factors, including the viral strain, cell type and assay used, and on adjustments made for plasma protein binding. Drug-resistant strains tend to have higher IC50 values compared to wild type.
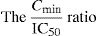

4. Protease inhibitor boosting
The goal of PI boosting is to increase the exposure to these agents, ensuring sustained and effective concentrations throughout the dosing interval. PI boosting is best achieved by administering ritonavir (usually low dose 100mg once or twice daily) along with the PI.
Ritonavir may increase exposure of a concomitantly administered PI by exerting the following effects:
(1)Ritonavir inhibits P-glycoprotein transport in the intestine, which increases absorption of the second PI. .
.
PK parameters..............................................Clinical advantages
Decreased systemic clearance(by ritonavir PK boosting)......Lower likelihood of resistance
Increased trough (Cmin)..................................Improved antiviral activity
Decreased peak (Cmax)...................................Reduced drug toxicity
Reduced pharmacokinetic variability............Amelioration of food restrictions
Increased AUC..................................................Improves adherence
4.1. PK of clinically tested STAT-C and ritonavir boosting
Pharmacokinetics parameters for boceprevir were evaluated on day 1 of monotherapy and on days 1 and 8 of combination therapy with PEG-IFN α-2b. The AUC on day 1 of monotherapy and combination therapy of both dose groups are similar, which suggests that there was no interaction between boceprevir and PEG-IFN.
HCV replication is characterized by a short virion half-life and a high production and clearance rate of free virions [24]. The estimated average half-life of the hepatitis C virus is 2.7h, with pretreatment production and clearance of 1012 virions per day. The HCV/NS5B RNA-dependent RNA polymerase has a low fidelity and no proofreading function.
From treatment of HIV and HBV with specific inhibitors, it is known that rapid selection of preexisting drug-resistant variants occurs during treatment. During selection pressure, the relative replication fitness of a selected drug-resistant variant determines whether the variant will grow out. Without selection pressure, a drug resistant variant may be generated but will simply vanish if its relative replication fitness is lower than that of non-drug-resistant variants.
The speed of selecting drug resistance depends mainly on the turnover of the viral nucleic acid; the HCV RNA strands present in infected hepatocytes serve as templates for producing new HCV virions that are released soon after [24]. As a result, the viral genetic half-life is shorter for HCV than for HIV and HBV and the time required for selecting drug-resistant mutants and for their expansion to become the major part of the viral population is shorter for HCV than for HIV and HBV. Thus, resistance to HCV antivirals is more likely than resistance to antiretrovirals [24], [25].
The presence of pre-existing mutations in treatment naïve patients may be a factor affecting the response to therapy. Indeed, the PI-resistant mutation R155K was recently detected as the dominant quasispecies in a treatment-naıve patient [34].
In a study by Kuntzen et al. dominant mutations were mostly observed as sporadic, unrelated cases at frequencies between 0.3% and 2.8% in the population. Taken together, however, 8.6% of patients infected with genotype 1a and 1.4% of patients infected with genotype 1b exhibited at least one drug resistance mutation, including two cases with possible multidrug resistance. Viral loads were high in the majority of patients carrying these mutations, suggesting that resistant viruses might achieve replicative capacities comparable to nonresistant strains in vivo [35].
Treatment success as measured by decreasing viral loads represents a balance between the achievable drug concentration in the plasma, the frequency and degree of drug resistance among the viral quasispecies [36], and the replication efficiency of drug-resistant viral strains. Numerous studies have investigated the impact of amino acid substitutions on viral replication and drug susceptibility to investigational NS5B polymerase or NS3/4A protease inhibitors in the replicon model. A pronounced reduction in the replicative capacity was described for the highly resistant A156 variants in NS3 and M423 variants in NS5B, and also to a lesser extent for the low-level PI-resistant R155, T54, and V36 mutations [11], [29], [33], [35]. It has been speculated that these lower replication levels could facilitate eradication through combination treatment [28], because STAT-C resistant viral strains appear to remain sensitive to interferon and ribavirin [33]. In a recent clinical trial, the in vivo relevance of these findings was supported by the observation that weakly telaprevir-resistant variants rose predominantly in patients who only achieved lower plasma drug levels, whereas increasing drug concentrations selected for mutants that were also highly resistant in vitro [11].
Preliminary results indicate that dominant resistance mutations can potentially reduce the early treatment response to STAT-C drugs [36], and it must be assumed that low-level resistant strains will sustain viral replication in patients who do not achieve optimal drug levels with standard dosing, in cases with poor adherence, or when dose reductions are inevitable due to adverse events. Importantly, continued viral replication in the presence of the selecting drug would put the patient at risk of developing additional resistance mutations. Here, observations from HIV infection suggest that baseline resistance even against only one drug in a multidrug antiviral regimen may affect treatment success [11], and further indicate that apart from single mutations conferring high level resistance, stepwise accumulation of subtle but synergistically acting resistance mutations may also eventually lead to treatment failure [37]. Data from a recent study using telaprevir has indicated a similar pathway in HCV infections, where combination of the low-level resistance mutations V36M and R155K resulted in a highly drug resistant phenotype, the appearance of which coincided with viral breakthrough [11], [28].
In HIV infection, resistance testing was calculated to be cost-effective when the prevalence of drug resistance becomes 1% at baseline and is currently recommended in areas with more than 5% prevalence of resistant strains, and in all cases of treatment failure [38]. For novel STAT-C drugs, important factors such as their cost and treatment response rates are not yet available to derive similar calculations, but further studies are needed to address these issues.
6. Tools for monitoring viral resistance
Characterizing resistance to STAT-C in clinical trials is essential for the management of a drug regimen to reduce the development of resistance and thereby maximize SVR rate. So far, methods to characterize viral resistance works in a complementary way and are distinct in genotypic and phenotypic assays [2].
6.1. Genotypic assays
Genotypic assays examine the genetic sequence of a target region involved directly or indirectly in the interaction of a drug with its target [11], [28], [39]. Initial characterization of the resistance profile for a drug requires comparing viral sequences before, during and after treatment to detect changes that occur during treatment. Available genotypic assays have different levels of sensitivity.
More sensitive methods such as clonal sequencing or the TaqMan™ mismatch amplification mutation assay (TaqMAMA) [40], may be more costly and time consuming.
Although not a measure of resistance per se, the viral fitness (replicative capacity) of resistant variants is an important factor, with implications for clinical resistance.
The replication capacity of HCV variants is typically assessed in vitro using a transient replicon system [7], or by comparing colony formation efficiency of the mutant replicon RNA with that of WT variants in co-culture growth competition assays.
Fitness has also been determined in vivo [11] by using HCV RNA levels and clonal sequencing to calculate the frequency of a given variant over time after the end of dosing to assess the growth rate compared with WT in the absence of drug-selective pressure.
In clinical research, viral variants identified by genotypic testing should be tested with a phenotypic assay, both to confirm that the mutation confers resistance to the drug and to assess the degree of resistance [41], [42], [43]. Phenotypic assays measure the IC50 in an enzyme or replicon assay. By testing the HCV variants for drug susceptibility in vitro, the fold change in sensitivity can be calculated as the IC50 value of the isolate/IC50 value of the reference strain (e.g. WT).
7. Identification of genotype/subtype
A major issue that limits the efficacy of NS3/4A protease inhibitors is the finding that genetic barrier and resistance profiles substantially differ between the different genotype 1 subtypes. The reason is that only one nucleotide substitution is needed to generate a subtype 1a sequence variant, whereas two substitutions are needed to the 1b sequence. In vivo, different resistance profiles in patients infected by HCV subtypes 1a and 1b have been demonstrated. In the former, the V36 and R155 substitutions represent the backbone of resistance, whereas in the latter resistance is less frequent as it is preferentially associated with substitutions at position A156 that are associated with a decreased fitness of the variants [10], [33], [41].
A correct identification of HCV subtypes 1a and 1b is hence crucial in clinical trials designated for new HCV drugs to avoid misinterpretation of efficacy and resistance data. It may also become important in future clinical practice, when therapy schedule needs to be tailored in genotype 1 patients according to genotype subtype. To identify the HCV genotype and subtype both in clinical trials and practice, commercial assays have been developed, most of them targeting the 59 noncoding region (59NCR) of the HCV genome [43].
As monotherapy, Direct Antiviral Agents (DAA) of several classes have been shown to induce significant viral suppression, with the addition of PEG-IFN/RBV or even PEG-IFN alone conferring additional viral suppression and/or markedly inhibiting the development of resistance to the direct-acting antiviral agent [28]. In the PROVE1 and PROVE2 studies of genotype 1 treatment-naive patients treated with the PI telaprevir, viral breakthrough was an infrequent event, especially in patients who experienced RVR [44], [45]. Similarly, in the SPRINT-1 boceprevir trial in treatment-naive patients, only a small number of patients discontinued for viral breakthrough [46]. These observations, together with in vitro experiments demonstrating the capacity of two or three drug combinations to induce additive or synergistic viral suppression [47], as well as the history of HIV therapy, provide a compelling case for the exploration of combinations of direct-acting antiviral agents. The ultimate goal of this approach is to build regimens able to overcome the development of resistance. As suggested in a interesting review by Pereira & Jacobson, the height of the “genetic barrier”, such as the relative likelihood to acquire conferring resistance mutations, varies among the different STAT-C drugs. Nucleoside analogues seem to have the highest barrier when compared to protease and non-nucleosides inhibitors. The combination of drug with different resistance profiles may become an interesting strategy to avoid the development of overlapping resistances [48]. So far, combinations have been studied with IFN as a cornerstone, but ultimately the development of IFN-free regimens is a major goal. Very recently, the INFORM-1 study, an intriguing dose-ranging, exploratory study of R7227, a PI, combined with R7128, a nucleoside polymerase inhibitor, for up to 2 weeks, demonstrated marked viral suppression [49]. No viral breakthroughs were reported. Data from the RBV-free arm of PROVE2 [47], the RBV-free arm of PROVE3, a study on telaprevir in earlier nonresponders [44] and the low dose RBV arm of SPRINT-1 [46] have provided a compelling case for the role of RBV in preventing the emergence of resistant variants. Such data provide a foundation for the early exploration of IFN-free regimens consisting of two DAA agents plus RBV. Given the universal interest in studying combinations of DAA agents, the timeline for the development of such combinations is a critical issue.
The success of STAT-C agents will depend on their ability to inhibit the replication of a broad range of viral quasispecies and prevent emergence of drug-resistant mutants. Present and forthcoming drugs should have a pharmacokinetic profile which could warrant plasma levels able to inhibit the viral replication along the inter-dose period. Moreover, treatment regimens based on the combination of drugs with different resistance profile may be the best strategy for improving the response rate in difficult to treat patients in the years to come.
The authors declare that they have no conflict of interest to disclose.
References
[1]. [1]World Health Organization. Viral cancers. Available at: http://www.who.int/vaccine_research/diseases/viral_cancers/en/index2.html [accessed 01.07.09].
[2]. [2]Kieffer TL, Kwong AD, Picchio GR. Viral resistance to specifically targeted antiviral therapies for hepatitis C (STAT-Cs). J Antimicrob Chemother. 2010;65(February (2)):202–212. CrossRef
[3]. [3]Ghany MG, Strader DB, Thomas DL, et al. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335–1374. CrossRef
[4]. [4]Lamarre D, Anderson PC, Bailey M, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. CrossRef
[5]. [5]Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol. 2010;105(May (5)):989–1004quiz 988, 1005. CrossRef
[6]. [6]Lohmann V, Korner F, Koch J, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. MEDLINE CrossRef
[7]. [7]Ludmerer SW, Graham DJ, Boots E, et al. Replication fitness and NS5B drug sensitivity of diverse hepatitis C virus isolates characterized by using a transient replication assay. Antimicrob Agents Chemother. 2005;49:2059–2069. MEDLINE CrossRef
[8]. [8]Shi ST, Herlihy KJ, Graham JP, et al. In vitro resistance study of AG-021541, a novel nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother. 2008;52:675–683. CrossRef
[9]. [9]Yi M, Tong X, Skelton A, et al. Mutations conferring resistance to SCH6, a novel hepatitis C virus NS3/4A protease inhibitor. Reduced RNA replication fitness and partial rescue by second-site mutations. J Biol Chem. 2006;281:8205–8215. MEDLINE CrossRef
[10]. [10]Tong X, Chase R, Skelton A, et al. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res. 2006;70:28–38. MEDLINE CrossRef
[11]. [11]Sarrazin C, Kieff er TL, Bartels D, et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology. 2007;132:1767–1777. Abstract Full Text Full-Text PDF (1908 KB) CrossRef
[12]. [12]De Francesco R, Carfi A. Advances in the development of new therapeutic agents targeting the NS3-4A serine protease or the NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Adv Drug Deliv Rev. 2007;59:1242–1262. CrossRef
[13]. [13]Thompson AJ, McHutchison JG. Review article: investigational agents for chronic hepatitis C. Aliment Pharmacol Ther. 2009;29(April (7)):689–705. CrossRef
[14]. [14]Thimme R, Lohmann V, Weber F. A target on the move: innate and adaptive immune escape strategies of hepatitis C virus. Antiviral Res. 2006;69:129–141. MEDLINE CrossRef
[15]. [15]Watashi K, Ishii N, Hijikata M, et al. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol Cell. 2005;19:111–122. MEDLINE CrossRef
[16]. [16]Ferguson NM, Fraser C, Anderson RM. Viral dynamics and anti-viral pharmacodynamics: rethinking in vitro measures of drug potency. Trends Pharmacol Sci. 2001;22(February (2)):97–100. MEDLINE CrossRef
[17]. [17]Griffin JP, O’Grady J, O’Grady J. The textbook of pharmaceutical medicine. 5th ed.. Blackwell; 2006;[Chapter 5].
[18]. [18]Pérez-Elías MJ, Morellon ML, Ortega E, et al. Pharmacokinetics of fosamprenavir plus ritonavir in human immunodeficiency virus type 1-infected adult subjects with hepatic impairment. Antimicrob Agents Chemother. 2009;53(December (12)):5185–5196. CrossRef
[19]. [19]Danner SA, Carr A, Leonard JM, et al. A short-term study of the safety, pharmacokinetics, and efficacy of ritonavir, an inhibitor of HIV-1 protease. European-Australian Collaborative Ritonavir Study Group. N Engl J Med. 1995;333(23):1528–1533. MEDLINE CrossRef
[20]. [20]Perni RB, Almquist SJ, Byrn RA, et al. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob Agents Chemother. 2006;3:899–909.
[21]. [21]Summa V. VX-950 (Vertex/Mitsubishi). Curr Opin Investig Drugs. 2005;8:831–837.
[22]. [22]Gentile I, Viola C, Borgia F, et al. Telaprevir: a promising protease inhibitor for the treatment of hepatitis C virus infection. Curr Med Chem. 2009;16(9):1115–1121Review. CrossRef
[23]. [23]Sarrazin C, Rouzier R, Wagner F, et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology. 2007;132(April (4)):1270–1278. Abstract Full Text Full-Text PDF (586 KB) CrossRef
[24]. [24]Soriano V, Perelson AS, Zoulim F. Why are there different dynamics in the selection of drug resistance in HIV and hepatitis B and C viruses?. J Antimicrob Chemother. 2008;62:1–4. CrossRef
[25]. [25]Kronenberger B, Zeuzem S. Treatment of chronic hepatitis C: anticipated impact of resistance in patients treated with protease inhibitors. Curr Gastroenterol Rep. 2009;11(February (1)):15–21. CrossRef
[26]. [26]Cubero M, Esteban JI, Otero T, et al. Naturally occurring NS3-protease-inhibitor resistant mutant A156T in the liver of an untreated chronic hepatitis C patient. Virology. 2008;370:237–245. CrossRef
[27]. [27]Lu L, Mo H, Pilot-Matias TJ, et al. Evolution of resistant M414T mutants among hepatitis C virus replicon cells treated with polymerase inhibitor A-782759. Antimicrob Agents Chemother. 2007;51:1889–1896. MEDLINE CrossRef
[28]. [28]Kieffer TL, Sarrazin C, Miller JS, et al. Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology. 2007;46:631–639. CrossRef
[29]. [29]He Y, King MS, Kempf DJ, et al. Relative replication capacity and selective advantage profiles of PI-resistant HCV NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob Agents Chemother. 2007;52:1101–1110. CrossRef
[30]. [30]Lu L, Pilot-Matias TJ, Stewart KD, et al. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob Agents Chemother. 2004;48:2260–2266. MEDLINE CrossRef
[31]. [31]Mo H, Lu L, Pilot-Matias T, et al. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an HCV serine protease inhibitor in vitro. Antimicrob Agents Chemother. 2005;49:4305–4314. MEDLINE CrossRef
[32]. [32]Yi M, Tong X, Skelton A, et al. Mutations conferring resistance to SCH6, a novel hepatitis C virus NS3/4A protease inhibitor. Reduced RNA replication fitness and partial rescue by secondsite mutations. J Biol Chem. 2006;281:8205–8215. MEDLINE CrossRef
[33]. [33]Zhou Y, Bartels DJ, Hanzelka BL, et al. Phenotypic characterization of resistant Val36 variants of hepatitis C virus NS3-4A serine protease. Antimicrob Agents Chemother. 2008;52:110–120. CrossRef
[34]. [34]Colson P, Brouk N, Lembo F, et al. Natural presence of substitution R155K within hepatitis C virus NS3 protease from a treatment-naive chronically infected patient. Hepatology. 2008;47:766–767. CrossRef
[35]. [35]Kuntzen T, Timm J, Berical A, et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naïve patients. Hepatology. 2008;48(December (6)):1769–1778. CrossRef
[36]. [36]Bartels DJ, Zhou Y, Zhang E, et al. Natural prevalence of HCV variants with decreased susceptibility to NS3-4A protease inhibitors in treatment-naive subjects. J Hepatol. 2008;48(Suppl. 2):S316. Full-Text PDF (55 KB) CrossRef
[37]. [37]Kuritzkes DR. Preventing and managing antiretroviral drug resistance. AIDS Patient Care STDS. 2004;18:259–273. MEDLINE
[38]. [38]Hirsch MS, Brun-Vezinet F, Clotet B, et al. Antiretroviral drug resistance testing in adults infected with human immunodeficiency virus type 1: 2003 recommendations of an International AIDS Society-USA Panel. Clin Infect Dis. 2003;37:113–128. CrossRef
[39]. [39]Qiu P, Sanfiorenzo V, Curry S, et al. Identification of HCV protease inhibitor resistance mutations by selection pressure-based method. Nucleic Acids Res. 2009;37:e74. CrossRef
[40]. [40]Curry S, Qiu P, Tong X. Analysis of HCV resistance mutations during combination therapy with protease inhibitor boceprevir and PEG-IFN alpha-2b using TaqMan mismatch amplification mutation assay. J Virol Methods. 2008;153:156–162. CrossRef
[41]. [41]Zhou Y, Müh U, Hanzelka BL, et al. Phenotypic and structural analyses of hepatitis C virus NS3 protease Arg155 variants: sensitivity to telaprevir (VX-950) and interferon alpha. J Biol Chem. 2007;282:22619–22628. CrossRef
[42]. [42]McCown MF, Rajyaguru S, Kular S, et al. GT-1a orGT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother. 2009;53:2129–2132. CrossRef
[43]. [43]Chevaliez S, Bouvier-Alias M, Brillet R, Pawlotsky JM. Hepatitis C virus (HCV) genotype 1 subtype identification in new HCV drug development and future clinical practice. PLoS One. 2009;4(December (12)):e8209. CrossRef
[44]. [44]McHutchison JG, Everson GT, Gordon SC, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009;360:1827–1838. CrossRef
[45]. [45]Hezode C, Forestier N, Dusheiko G, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 2009;360:1839–1850. CrossRef
[46]. [46]Kwo P, Lawitz E, McCone J, et al. HCV SPRINT-1: final results: SVR 24 from a phase 2 study of boceprevir plus PegIntron (Peginterferon alfa-2b)/ribavirin in treatment-naïve subjects with genotype-1 chronic hepatitis C. J Hepatology. 2009;50(Suppl. 1):S4.
[47]. [47]Grunberger C, Wyles DL, Kaihara KA, et al. 3-Drug synergistic interactions of small molecular inhibitors of hepatitis C virus replication. J Infect Dis. 2008;197:42–45. CrossRef
[48]. [48]Pereira A, Jacobson IM. New and experimental therapies for HCV. Nat Rev Gastroenterol Hepatolol. 2009;6:403–411.
[49]. [49]Gane EJ, Roberts SK, Stedman C, et al. First-in-man demonstration of potent antiviral activity with a nucleoside polymerase (R7128) and protease (R7227/ITMN191) inhibitor concentration in HCV: safety, pharmacokinetics, and virologic results from INFORM-1. J Hepatology. 2009;50(Suppl. 1):S380.
Department of Infectious Diseases, Foundation IRCCS San Matteo Hospital - University of Pavia, Pavia, Italy
Corresponding author at: Department of Infectious Diseases, Hepatology Outpatients Unit, University of Pavia, Fondazione IRCCS Policlinico San Matteo, Via Taramelli, 5, 27100 Pavia, Italy. Tel.: +39 0382 501080; fax: +39 0382 501080.
PII: S1590-8658(10)00314-2
doi:10.1016/j.dld.2010.09.007
© 2010 Published by Elsevier Inc.
No comments:
Post a Comment